Synthesis of 2,3-MethylenedioxyamphetaminesForens. Sci. Int. 114, 139-153 (2000)[ Back to the Chemistry Archive ] Introduction Some phenylethylamines such as 1-(3,4-methylenedioxyphenyl)-2-propanamine derivatives are increasingly abused psychoactive drugs and well documented in the literature [1,2]. The most popular compound of the various clandestine products is N-methyl-3,4-methylenedioxyamphetamine (MDMA), also known as Ecstasy. The continuing designer drug exploration of the homologous series and their widespread consumption results in an increasing number of reports regarding abuse and intoxication. Some of these novel 3,4-methylenedioxyphenylamphetamines have a complete different psychoactive spectrum [3] compared to the classical hallucinogenic amphetamines and rather induce facilitated communication and introspective states. These compounds were described as representing a new pharmacological class, the entactogens. Phenylethylamine derivatives with a 1-(3,4-methylenedioxyphenyl)-2-butanamine structure produce emphasizing and enhancing effects on empathy without any sympathomimetic side effects of the corresponding amphetamines [4]. The preparation of the 2,3-butanamine derivatives (3a-3d) have not been described previously [5]. The general synthetic scheme for the preparation of the 2,3-(methylene- dioxyphenyl)-2-butanamine isomers (3a-3d) is presented in the scheme below. In summary, 2,3-Methylenedioxybenzaldehyde was condensed with nitropropane to give ß-ethyl-2,3-methylene- dioxy-ß-nitrostyrene. Reduction with LiAlH4 gave 1-(2,3-methylenedioxyphenyl)-2-butanamine (3a). Formylation, acetylation and subsequent reduction turned it to N-methyl-1-(2,3-methyl- enedioxyphenyl)-2-butanamine (3b) and N-ethyl-1-(2,3-methylenedioxyphenyl)- 2-butanamine (3c). N,N-dimethyl-1-(2,3-methylenedioxyphenyl)-2-butanamine (3d) was prepared from 3a by reduction of the formimine and the following formylation and reduction of the N-methylformamid. All compounds were isolated as their hydrochloride or perchlorate salts. The preparation of N-alkylated amines followed a trend of decreasing yields: The primary amine 1-(2,3-methylenedioxyphenyl)-2-butanamine (3a) = 82%, the secondary N-methyl analog (3b) = 41%, the N-ethyl = 53% and the N,N-dimethyl (3d) = 5%; the results suggest that steric hindrance plays a significant role in the overall stabilities of N-substituted compounds. 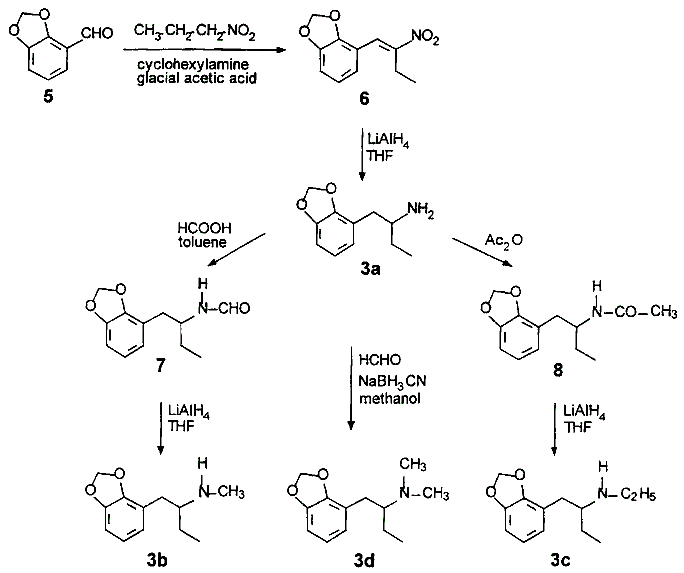
ß-ethyl-2,3-methylenedioxy-ß-nitrostyrene (6) 2,3-Methylenedioxybenzaldehyde (5) (3.0 g, 22 mmol), nitropropane (3.0 g, 34 mmol) and cyclohexylamine (2.2 g, 22 mmol) were dissolved in 20 ml of glacial acetic acid and heated to 95°C. After 6 h, the hot, yellow-brown solution was poured on crushed ice, stirred and acidified with concentrated HCl, whereby the crude product precipitated. Recrystallization from isopropanol provided 2.3 g (47% yield) of the nitrostyrene as bright yellow crystals. mp: 62°C. 1-(2,3-methylenedioxyphenyl)-2-butanamine hydrochloride (3a.HCl) A solution of 2,3-methylenedioxy-ß-ethyl-ß-nitrostyrene (3.7 g, 17 mmol) in 20 ml of THF was added for 30 min to a solution of LiAlH4 (4.2 g, 110 mmol) in 50 ml of THF and then heated to reflux. After 5 h, the excess LiAlH4 was quenched with dropwise water and the precipitated inorganic salts were removed via suction filtration. The filtrate was evaporated in vacuo to a yellow oil which was treated with etheral HCl and white crystals precipitated to give 2.8 g (85% yield) of 3a.HCl. mp: 168°C. N-formyl-1-(2,3-methylenedioxyphenyl)-2-butanamine (7) A solution of 3a (2.24 g, 12 mmol) and 1.0 ml of formic acid 98% (1.0 g, 22 mmol) were added to 100 ml of toluene and heated to reflux with a soxhlet containing 4 Ĺ molecular sieve. After 48 h the resulting solution was evaporated in vacuo to give a colored oil, which was dissolved in 100 ml of dichloromethane and washed several times with diluted HCl. The organic layer was dried over anhydrous sodium sulfate and evaporated in vacuo to give 2.1 g (82% yield) of N-formyl-1-(2,3-methylenedioxyphenyl)-2-butanamine as a yellow oil. N-methyl-1-(2,3-methylenedioxyphenyl)-2-butanamine hydrochloride (3b.HCl) A solution of 7 (2.0 g, 9 mmol) in 10 ml of THF was added over 15 min to 60 ml of a solution of 2.28 g LiAlH4 in THF and heated to reflux. After 4.5 h the excess LiAlH4 was quenched dropwise with water in the usual manner. The oil was dissolved in dilute HCl and washed with dichloromethane, alkalized with concentrated NaOH and then extracted with dichloromethane. The combined organic layers were dried over anhydrous sodium sulfate and evaporated in vacuo to a light yellow oil, which was diluted with etheral HCl to give 0.8 g (41% yield) 3b.HCl mp 139°C. N-ethyl-1-(2,3-methylenedioxyphenyl)-2-butanamine perchlorate (3c.HClO4) A solution of 8 (1.0 g, 4.3 mmol) in 10 ml of THF was added for 5 min to a solution of LiAlH4 (0.6 g,15.8 mmol) in 20 ml of THF and heated to reflux. After 3.5 h the excess LiAlH4 was quenched in the usual manner. The extracted oil was evaporated in vacuo to a light yellow oil, which was dissolved in a solution of isopropanol, concentrated perchloric acid and diethyl ether (3:1:20) to get a spontaneous crystallization of 0.5 g (53% yield) 3c.HClO4. mp: 126°C. N-acetyl-1-(2,3-methylenedioxyphenyl)-2-butanamine (8) A solution of 3a (1.0 g, 5.2 mmol) in 2 ml of acetic anhydride (1.16 g, 11 mmol) was heated in a 10 ml glass tube to 75°C with continuous stirring. After 24 h the cooled mixture was dissolved in 30 ml of dichloromethane, washed several times with dilute HCl, dilute NaOH, water and then dried over anhydrous sodium sulfate. Evaporation under vacuum afforded 1.0 g (82% yield) of N-acetyl-1-(2,3-methylene-dioxyphenyl)-2-butanamine as an amber solid. mp: 93°C. N,N-dimethyl-1-(2,3-methylenedioxyphenyl)-2-butanamine perchlorate (3d.HClO4) A solution of 3a (l.2 g, 6.2 mmol) and 6.12 ml of 35% formaldehyde (6.67 g, 60 mmol) in 30 ml of methanol was stirred at room temperature. Sodium cyanoborohydride (2.5 g, 40 mmol) was added, the reaction mixture was maintained at pH 6 by adding concentrated HCl dropwise and then heated to reflux for 48 h. The mixture was suspended with water, acidified with dilute HCl and washed with dichloromethane, then alkalized with concentrated NaOH and extracted with dichloromethane. The combined extracts were dried over anhydrous sodium sulfate and evaporated in vacuo to a light yellow oil, which was dissolved in a solution of isopropanol, concentrated perchloric acid and diethyl ether to get a spontaneous crystallization of 0.1 g (5% yield) 3d.HClO4. mp:101°C. References [1] T.A. Dal Cason, MDA Analogs and Homologs, Forensic Sci. Int. 35, 675 (1990)
|