Abstract
The copper(I)-catalyzed exchange of bromine by methoxide in 5-bromovanillin and in 3,5-dibromo-4-hydroxybenzaldehyde provides an efficient method for preparing 3,5-dimethoxy-4-hydroxybenzaldehyde, from which 3,4,5-trimethoxybenzaldehyde is obtained by methylation with dimethylsulfate.
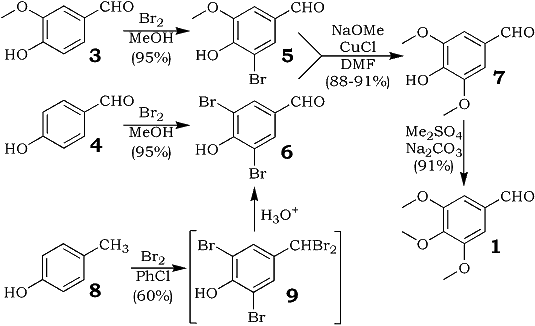
3,4,5-Trimethoxy- benzaldehyde (1) is an important starting material used in the synthesis of certain pharmaceuticals, such as the antibacterial trimethoprim (2)1.
Previous syntheses of 1 have been achieved from 3,4,5-trimethoxybenzoyl chloride by a modified Rosenmund reduction2 and by application of the Reissert reaction3. Although both routes are satisfactory for most purposes, certain considerations, primarily economic, led us to examine other approaches to 1.
We describe herein the preparation of 1 that relies on the preparation of syringaldehyde (7) from 5-bromovanillin (5) and from 3,5-dibromo-4-hydroxybenzaldehyde (6) using a copper(I)-catalyzed exchange of bromine by methoxide in DMF4. The high yields obtained (88-91%) by us for the exchange reaction is somewhat surprising since Bacon and Rennison5 had reported that the presence of an aldehyde group is deleterious to the reaction. However, it should be noted that they had carried out the exchange reaction in the presence of 2,4,6-collidine.
Bromination of vanillin (3) in methanol gave 5-bromovanillin (5) directly from the reaction medium in 95% yield. Treatment of 5 in DMF with ca. four equivalents of freshly prepared sodium methoxide in the presence of a catalytic amount of cuprous chloride6 gave, after acidification, syringaldehyde (7) in 91% yield. Methylation of 7 with dimethylsulfate in the presence of Na2CO3 gave 1 in 91% yield. In a similar manner, 4-hydroxybenzaldehyde was brominated to give 3,5-dibromo-4-hydroxybenzaldehyde (6), which was converted into 7 in 88% yield by the copper(I)-catalyzed exchange of the bromo substituents by methoxide.
3,5-Dibromo-4-hydroxy-benzaldehyde was also prepared by the direct bromination of p-cresol with 4.5 equivalents of bromine in chlorobenzene, initially at 25°C and then at reflux, to give the tetrabromide 9, hydrolysis of which with 1N HCl afforded 6 in 60% yield. The major by-products formed in this synthesis of 6 were the ester 10 (20%) and the ether 11 (7%).
Experimental
General
Melting points were determined in capillaries on a Thomas-Hoover melting point apparatus and are uncorrected. Unless otherwise indicated, infrared (IR) and nuclear magnetic resonance spectra (NMR) were determined in CHCl3 and CDCl3, respectively; 1H and 13C NMR spectra were recorded at 200 and 50.4 MHz, respectively. Chemical shifts are expressed in parts per million (ppm) relative to tetramethylsilane, and coupling constants (J) are expressed in hertz (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet). Mass spectra (MS) were determined with a direct inlet system with ionization energy of 70 eV; m/z values are given with relative intensities (%) in parentheses. Thin-layer chromatograms (TLC, silica gel G) were purchased from Merck (Darmstadt); spots were visible under short wavelength UV light or made visible by spraying with 10% phosphomolybdic acid in ethanol and heating the plates to 100°C.
5-Bromovanillin (5)
To a stirred, cooled (0°C) solution of 152.15 g (1.0 mol) of vanillin in 1.0 L of methanol was added during 20 min 176.0 g (1.1 mol) of bromine at such a rate that the temperature was kept below 20°C. The mixture was stirred at room temperature for 1 h, cooled to 0°C, and treated during 30 min with 500 mL of cold (5°C) water. Stirring was continued for 15 min and the product was collected by filtration. It was washed with water (4 x 500 mL), then with 500 mL of cold (0°C), 70% methanol, and dried in vacuo at 50°C overnight to give 218.5 g (95%) of 5 as pale yellow crystals, mp 163-164°C (Lit.7 mp 163-164°C);
Syringaldehyde (7)
- From 5-Bromovanillin (5)
Under a blanket of argon, a solution of 214 g (0.93 mol) of 5-bromovanillin in 420 mL of anhydrous DMF was added during 25 min to a stirred slurry of freshly prepared, powdered sodium methoxide (3.72 mol, from 85.2 g of clean sodium in 1.0 L of CH3OH, followed by removal of the excess CH3OH by distillation under vacuum) and 10.7 g (0.054 mol) of freshly prepared cuprous chloride in 150 mL of anhydrous DMF. The mixture was stirred at 97°C for 2.5 h, whereupon the color changed from blue to green after 30 min, and then to beige after 45 min. It was cooled to 60°C, and the DMF was evaporated under vacuum (0.2 Torr). To the residue was added 1.0 L of 15% brine and the mixture was stirred at 50°C for 30 min, cooled to 0°C and slowly acidified with 300 mL of cold (0°C) conc. hydrochloric acid. Stirring was continued at room temperature for 1.0 h and the crude product was collected by filtration. It was washed with 4 x 400 mL of cold H2O until neutral and extracted with warm (60°C) ethyl acetate (5 x 500 mL). The extract was evaporated to give 154 g (91%) of syringaldehyde, mp 109-111°C (Lit.8 109-110°C); TLC (60:30:1 ethyl acetate:hexane:formic acid) showed only one spot, Rf 0.55; 5 had Rf 0.70.
Syringaldehyde prepared above may be crystallized from ethyl acetate (93% return) to give pale yellow crystals, mp 110-112°C. - From 3,5-Dibromo-4-hydroxybenzaldehyde (6)
A 165-g (0.59 mol) batch of 6 in 156 mL of methanol and 312 mL of anhydrous DMF was reacted during 4.0 h with 4.78 moles of freshly prepared sodium methoxide in the presence of 9.45g (0.047 mol) of cuprous chloride under the conditions described previously for the conversion of 5 to give 153.3 g (88% yield) of 7, mp 105-108°C.
3,4-Dibromo-4-hydroxybenzaldehyde
- From 4-Hydroxybenzaldehyde
To a stirred, cooled (0°C) solution of 122.0 g (1.0 mol) 4-hydroxybenzaldehyde in 1.0 L of methanol was added during 30 min 325.0 g (2.2 mol) of bromine at such a rate that the temperature was kept below 20°C. The mixture was stirred at room temperature for 1.0 h, 800 mL of methanol was removed by distillation (water aspirator) at 50°C, and to the warm (45°C) solution was added 2.0 L of water during 20 min. The mixture was stirred at 0°C for 1.0 h and the product was collected by filtration. It was washed with water (5 x 1.0 L) and then with 500 mL of cold, 30% aqueous methanol, dried in vacuo at 70°C for 18 h to give 264.4 g (95.5%) of 6 as a colorless powder, mp 180-182°C. TLC (silica gel, 30:60:1 ethyl acetate:hexane:formic acid) showed only one spot under short wavelength UV light. - From p-Cresol
A cooled (10°C), stirred solution of 108.1 g (1.0 mol) of p-cresol in 500 mL of chlorobenzene was treated during 30 min with 720 g (4.55 moles) of bromine in 600 mL of chlorobenzene at such a rate that the temperature was kept below 25°C. The mixture was then stirred at reflux for 4.5 h and concentrated in vacuo at 60°C to give 219 g of a red oil. This was dissolved in 500 mL of methanol, cooled to 5°C, and treated with 500 mL of 1.0N hydrochloric acid. The mixture was stirred at 5°C for 4.5 h, diluted with 1.0 L of cold water, and the product was collected by filtration. It was washed with 500 mL of cold 50% aqueous methanol and dried in vacuo at 35°C overnight to give 168.3 g (60%) of 6, mp 179-182°C.
3,4,5-Trimethoxybenzaldehyde (1)
A stirred mixture of 153.0 g (0.84 mol) of syringaldehyde (7) in 1.0 L of acetone, was treated with 127.0 g (1.02 mol) of dimethylsulfate, 118.0 g (1.11 mol) of anhyd. Na2CO3, and 3.0 g (0.053 mol) of KOH in 32 mL of water. The mixture was stirred under reflux for 18 h. cooled to room temperature and filtered. The filter cake was washed with acetone (3 x 200 mL) and the filtrate and washings were concentrated to ca. 200 mL. The stirred solution was diluted with 500 mL of water during 30 min, followed by 1.5 L of water during 15 min. Stirring was continued at room temperature for 15 min and then at 0°C for 30 min, and the product was collected by filtration. It was washed with water (4 x 2.0 L) and dried in vacuo at 25°C for 24 h to give 150.2 (91.5%) of 1 as colorless crystals, mp 73-75°C (Lit.9 74°C). TLC (4:1 ether-hexane) revealed only one spot, Rf 0.57.