Abstract
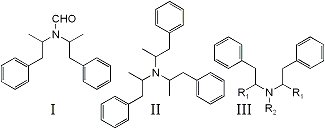
bis-(1-phenylisopropyl)formamide (I) has been identified as a by-product in the Leuckart synthesis of N-formylamphetamine1,2, a reaction precursor to DL-amphetamine. Significantly, it has been detected as an impurity in exhibits of illicitly manufactured DL-amphetamine3,4 but identified incorrectly as tri-(1-isopropylphenyl)amine (II)3.
Discussion
Compound I is detected as a doublet in the gas chromatographic analysis of impure N-formylamphetamine and DL-amphetamine, both peaks presenting identical mass spectra. The ions of greatest abundance appear at m/e 91 (base peak), 190, and 119. No ions are detected beyond m/e 190. The overall fragmentation pattern suggests a structure of the type shown as III in Fig. 1. Several representative compounds, some of which have been identified as impurities in illicit methamphetamine5-8, have been compared previously5. Mass spectral-structural correlations via the fragmentation processes outlined below are shown in Table 1:
- Cleavage beta to N9,10
- Fragmentation of ion produced by (a) alpha to N with charge transfer to the phenylalkyl moiety5,8.
- Alpha, beta cleavage to N with hydrogen rearrangement9,10; and
- Benzyl is indicated by the presence of an intense m/e 91 fragment with additional fragmentation at m/e 77, 65, 51, and 3911.
Since Mechanism (a) is expected to produce ions of great intensity in phenethylamines9,12,13, the lack of a m/e 280 fragment eliminates Structure II from serious consideration, particularly since its structure provides three sites for such cleavage to occur.
The great abundance of ions at m/e 119 and corresponding lack of m/e 105 fragments support the phenylisopropyl configuration for both major attachments to N. R3 must then consist of a group with a mass of 29. This may be attributed to either CH, CH3 or CHO. However, the amine function is all but denied by failure of the compound to extract with acid (dilute tartaric, hydrochloric, and sulfuric) to any detectable extent from ether.
Table I - Comparative data for major ions, for compounds of structural Type III
| m/e of the Ion Produced | |||||||
| Compound | Mol.Wt. | R1 | R2 | (a) | (b) | (e) | (d) |
| N-Methyl-diphenethylamine | 239 |
H | CH3 | 148 |
105 |
44 |
91 |
| bis -(1-Phenylisopropyl)amine | 253 |
CH3 | H | 162 |
119 |
44 |
91 |
| bis-(1-Phenylisopropyl)methylamine | 267 |
CH3 | CH3 | 176 |
119 |
58 |
91 |
| bis-(1-Phenylisopropyl)formamide (proposed) | 281 |
CH3 | CHO | 190 |
119 |
72 |
91 |
A doublet obtained by reverse phase high pressure liquid chromatography4 of an impure N-formylamphetamine synthesis product was found to correspond to the gas-liquid chromatography (GLC) doublet, both components eluting in the same order, as determined by gas chromatographic/mass spectral analysis of collected fractions. This result supported a hypothesis that the GLC doublet arises from stereoisomerism inherent the injected material4 and not from some form of chromatographic degradation. Rotational isomerism is also excluded as a factor.
Chloroform extracts of fractions representing pure cuts of each peak were evaporated and then subjected to infrareds and proton magnetic resonance analysis. The infrared spectra of both extracts showed no significant differences. The strongest band, at 1668 cm-1, confirmed the presence of carbonyl, suggesting, particularly, a tertiary amide. Lack of "Amide II" bands denied the presence of primary and secondary amide functions14. The second and third most intense bands, at 696 and 741 cm-1, indicated mono-substituted phenyl. Other bands (of medium intensity) appeared at 1494, 1453, 1431, 1375, 1310, 1270, 1150 (doublet), 1120, and 1028 cm-1.
Proton magnetic resonance spectroscopy (deuterated chloroform solution) clearly supported Structure I while emphasizing the stereoisomeric differences of the material. Assignments for the substance producing the earlier eluting chromatographic peak, obtained from chemical shifts, splitting patterns, and peak areas, are as follows: singlet at 8.22 ppm (one formyl hydrogen), broad peak at 6.9 to 7.5 ppm (ten phenyl hydrogens), multiplet at 3.93 ppm (one methine hydrogen), multiplet at 3.49 ppm (one methine hydrogen), doublets for the methylene hydrogens at 2.88 (one hydrogen), 2.83 (one hydrogen), and 2.55 ppm (two hydrogens), and doublets of three hydrogen intensity each st 1.21 and 1.10 ppm (methyl). Significant differences in the proton magnetic resonance spectrum of the other substances were evident for methine (multiplets of one hydrogen intensity each at 4.04 and 3.60 ppm) and methylene (doublets of one hydrogen intensity each at 3.03, 2.98, 2.78, and 2.75 ppm). The methyl protons (doublets of three hydrogen intensity, each) absorbed at 1.21 and 1.00 ppm. A more detailed report concerning the stereochemistry of Structure I is in process.
Conclusion
The synthesis of amphetamine by the Leuckart synthesis requires the reaction of methyl benzyl ketone with either formamide or ammonium formate, producing N-formylamphetamine as an intermediate1,2. It is believed that Structure I is produced by reaction between N-formylamphetamine and excess ketone. Its presence in DL-amphetamine, therefore, should provide strong evidence for the implication of the Leuckart synthesis in the manufacturing process.
Summary
An impurity previously reported in illicit amphetamine has been found as a by-product in the synthesis of N-formylamphetamine, a reaction precursor to amphetamine by the Leuckart synthesis. Its reidentification as bis-(1-phenylisopropyl)formamide has been supported by combined spectroscopic analysis of isolated fractions.