Abstract
A new environment friendly procedure for effective aromatic iodination is presented. A mixture of potassium iodide and potassium iodate is used in the presence of an acid for in situ iodination of aromatic compounds.
Aromatic iodo compounds are an important class of compounds in synthetic organic chemistry. They are useful for the preparation of organometallic reagents and some are potential intermediates for the synthesis of pharmaceutical and bioactive materials. They are useful in metal catalyzed coupling reactions which are widely applied in the preparation of complex molecules.
The iodination of organic molecules using elemental iodine is particularly sluggish compared to chlorination and bromination and needs activation for effective electrophilic substitution. The reagents reported for iodination of aromatic substrates include, N-iodosuccinimide (NIS),1 NIS-CF3SO3H,2 NIS-CF3COOH,3 I2-tetrabutylammonium peroxydisulfate,4 I2-nitrogen dioxide,5 I2-diiodine pentoxide6 I2-silver sulphate,7 I2-mercury salts,8a,b,c I2-chromium oxide,9 I2-lead acetate,10 I2-thallium acetate,11 I2-F-TEDA-BF4,12 iodine monochloride,13 BuLi-F3CCH2I,14 NaOCl-NaI,15bis(symcollidine)iodine(I) hexafluorophosphate,16 bis(pyridine)iodonium(I) tetrafluoroborate-CF3SO3H17 and NaI-ammonium hexanitrocerate.18 Most of these reagents are complicated, costly or use toxic heavy metal catalysts with potential environmental problems due to the generation of hazardous waste. In this note we wish to report our preliminary studies on an efficient, new environmentally benign procedure for aromatic iodination.
Table 1.
Iodination for aromatic compounds with KI/KIO3/H+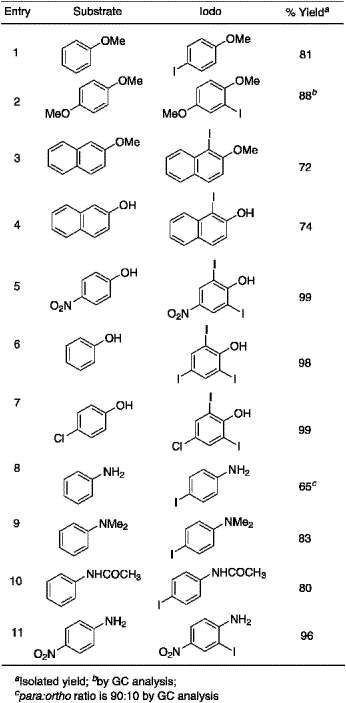
The oxidation of an iodide anion results in the formation of an electrophilic iodonium species which is responsible for iodination of the aromatic nucleus. Our method is based on this concept where a mixture of 0.66 mol equiv. of potassium iodide and 0.33 mol equiv. of potassium iodate is treated with one mol equiv. of a mineral acid. This results in the in situ formation of an iodonium cation which can effect aromatic iodination, see Eq. 1:
The test reaction was carried out on anisole in aqueous methanol with potassium iodide and potassium iodate in the presence of hydrochloric acid under ambient conditions. Careful analysis (TLC and GLC) of the reaction products confirmed the formation of 4-iodoanisole with excellent conversion and isolated yield. Having established the feasibility of this conversion, we undertook a systematic study and the results are summarized in Table 1.
Controlled mono iodination of 1,4-dimethoxybenzene was carried out with one equivalent of the iodinating reagent and the desired product, 2-iodo-1,4-dimethoxybenzene was formed in high yield. Iodination of 2-naphthol and 2-methoxynaphthalene resulted in 1-iodo derivatives as the sole products in good, isolable yields. Diiodination of 4-nitrophenol and 4-chlorophenol was achieved in excellent yields while triiodination of phenol was effected with three equivalents of the reagent. Iodination of aniline gave a mixture of para- and ortho-iodoaniline in the ratio of 90:10 in moderate yield, while 4-nitroaniline furnished the 2-iodo-compound in good yield. A previously reported procedure15 for aromatic iodination describes a very low yield of 4-iodo-N,N-dimethylaniline from N,N-dimethylaniline. It is remarkable to observe that the present reagent system was very effective for this particular substrate resulting in an excellent yield of 4-iodo-N,N-dimethylaniline. The iodinated products reported in this study were isolated by column chromatography over silica gel and characterized by standard analytical means and by comparison of their melting points with those reported.
Thus, we have reported our findings on the use of a simple reagent system for an environmentally benign and efficient procedure for the iodination of aromatic compounds.
Experimental
General procedure for the iodination:
4-Iodoanisole: A solution of anisole (1.00 g; 9.26 mmol), potassium iodide (1.03 g; 6.22 mmol) and potassium iodate (0.66 g; 3.08 mmol) was prepared in methanol (5 mL) and water (30 mL). This mixture was treated at room temperature with dilute hydrochloric acid (9.5 mmol) over 40 to 45 minutes and stirred for an additional 2?3 h, diluted with water (50 mL) and extracted with dichloromethane (25 mL?3). The combined organic extract was washed with dilute sodium thiosulphate (5%), water, brine, dried over anhydrous sodium sulphate and concentrated under reduced pressure to give a thick oil (1.94 g; 90%). Further purification was carried out by crystallization from cold hexane to afford a white crystalline product (1.75 g; 81%), mp 52?54?C (Lit.19 53?54?C), which showed satisfactory analytical and spectroscopic properties.