Abstract
An easy method for epoxidation of olefins using bleach (sodium hypochlorite) and either a stoichiometric or catalytic amount of bromide ion has been developed. Without any transition metal catalyst a variety of non-activated olefins give epoxides in high yields and good selectivity at ambient conditions.
Introduction
Oxidation reactions of olefins to give epoxides are of major importance for organic synthesis. Nowadays, especially asymmetric epoxidation reactions are in the focus of methodological developments1. However, the synthesis of racemic epoxides is still important on laboratory as well as industrial scales. A convenient method for the synthesis of epoxides is the oxidation of olefins with hydrogen peroxide or alkyl peroxides in the presence of transition metal complexes2. However, in general the activity of the catalyst is limited and the metal catalyst as well as modifying ligands have to be separated after the reaction. Nevertheless, significant advances have been made in non-asymmetric metalcatalyzed epoxidation reactions in the last decade3. Especially noteworthy with respect to simplicity and catalyst productivity was the development of redox active polyoxometalates (POM's) in combination with phase transfer active agents as catalysts in combination with hydrogen peroxide.
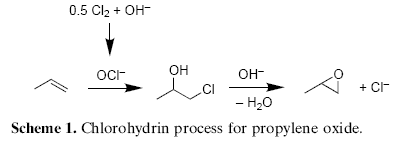
More traditionally, epoxides are synthesized by the reaction of olefins with hydrogen peroxide in the presence of acetic or formic acid4. This convenient method involves the in situ formation of the corresponding peracid, which easily undergoes epoxidation reaction. A drawback of this method are potential side reactions of the acid. Hence, the method is only of limited use for acid-labile olefins or epoxides. An alternative cheap and practical oxidant is bleach (sodium hypochlorite), which might be used either directly or is produced in situ from chlorine under basic conditions. Although in situ generated hypochlorite is still used in the two-step commercial process for propylene epoxide (Scheme 1)5, comparably few studies described the direct epoxidation of non-activated olefins with hypochlorite without metals being present6.
Some time ago we became interested in the improvement of known oxidation reactions of olefins. After having developed a new osmium-catalyzed dihydroxylation reaction using air as terminal oxidant7, we studied selective alcohol oxidation reactions8 and the catalytic dihydroxylation in the presence of sodium hypochlorite9 as oxidant. Based on this work, we turned interest to epoxidation reactions applying sodium hypochlorite. In this manuscript we describe a novel general method for the epoxidation of olefins using sodium hypochlorite in the presence of a catalytic or stoichiometric amount of bromide ion.
Results and Discussion
While investigating the oxidation of alpha-methylstyrene in the presence of different metal catalysts and sodium hypochlorite, we discovered that epoxidation to 2-phenyl-1-epoxypropane proceeds as a side-reaction independent from the metal catalyst used (Scheme 2).
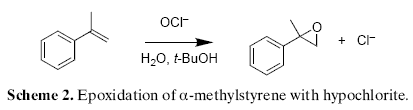
After studying the available literature we were surprised that the direct epoxidation of non-activate dolefins using sodium hypochlorite as oxidant has not been examined in more detail. Therefore, we decided to take a closer look at this reaction. As shown in Table 1 (Entry 1) the reaction proceeds in 15% yield using simple sodium hypochlorite in a biphasic mixture of water and tert-butyl alcohol at room temperature. We thought that the in situ generation of the more active hypobromide will increase the epoxide yield. Indeed, upon addition of 1.5 equiv. of KBr (with respect to sodium hypochlorite) the reaction proceeds smoothly within 2 h giving the corresponding epoxide in 90% yield at 25°C. Longer reaction times lead to slight decomposition of the desired product. The solvent system is of major influence for the outcome of the reaction. Using tert-butyl alcohol or water alone, the epoxide is obtained only in 1-4%. Biphasic mixtures of organic solvents and water give better results, however the yields with dichloromethane or tetrahydrofuran are still comparably low (ca. 30%). Mixtures of acetonitrile or tert-butyl alcohol and buffered water solution (pH=10.4) lead to the best epoxide yield (up to 90%). Variation of the pH from 9.5-12.0 and changing the reaction time do not have a significant influence on the reaction. In general, most of the reactions are finished after 0.5 h. Slightly lower yields are obtained at pH 11.6 and 12.
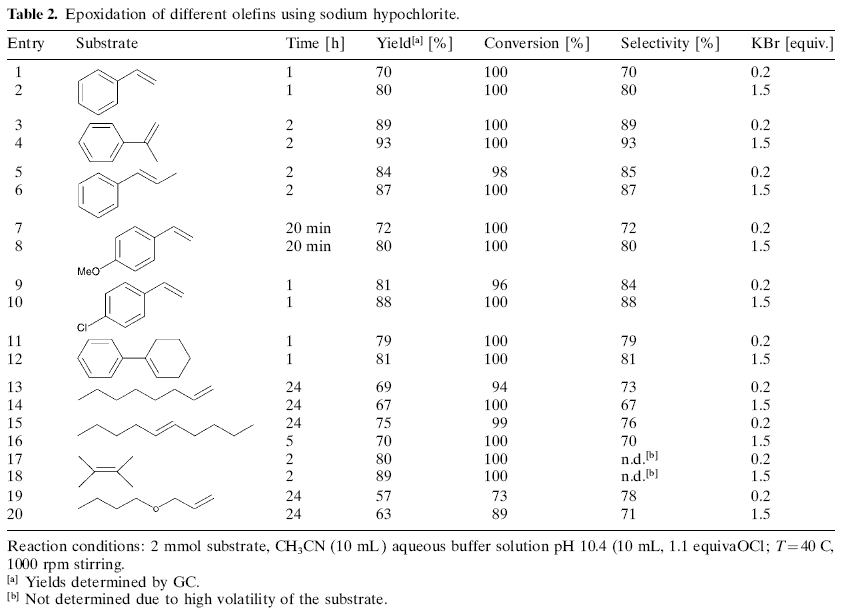
Advantageously, the epoxidation reaction with sodium hypochlorite also proceeds in the presence of catalytic amounts of bromide ions. The reduction of the amount of bromide ions from 1.5 equiv. to 0.2 equiv. leads only to a slight decrease of epoxide. Further reduction of the bromide concentration results in lower epoxide yield (46% at 0.05 equivalents of KBr). Next, we tested whether a combination of hydrogen peroxide and bromide ions is also able to effect epoxidation reactions. However, no conversion of α-methylstyrene is observed under these conditions. In order to get more information about the mechanism, we studied the concentration-time dependence of the olefin and reaction products via GC. A >95% conversion of α-methylstyrene is observed within the first minute. At the same time 2-phenyl-2-hydroxy-1-propyl bromide is formed in nearly 90% yield. This bromohydrin is converted immediately to the desired epoxide. While nearly 75% of the desired epoxide is obtained within 5 minutes, a maximum yield of epoxide (83%) is seen in between 30 and 60 minutes. Next, we studied the scope and limitations of the procedure. Different types of olefins were tested (Table 2). Aromatic olefins such as styrene, α-methylstyrene, β-methylstyrene, p-chlorostyrene, p-methoxystyrene, and 1-phenylcyclohexene give the corresponding epoxide in 70-93% yield (Table 2, entries 1-12). In general, the reaction is finished within 1 to 2 hours. Much longer reaction times can lead to slightly lower yields due to subsequent decomposition of the epoxide. In case of aromatic olefins apart from the desired epoxidation reaction small amounts of halogenation of the aromatic nucleus can be observed. In addition, the 1,2-dibromo or 1,2-chlorobromo derivatives arising from halogen addition along the double bond are detected. The slow increase in product selectivity with increased reaction time using α-methylstyrene as substrate (Table 2, entries 3 and 4) arises from the slow hydrolysis of the byproduct 1,2-dibromo-2-phenylpropane. This subsequent hydrolysis reaction is evident with all substrates forming benzylic bromides as byproducts; e.g., all substituted styrenes. All reactions proceed also well in the presence of catalytic amounts of bromide. However, the addition of 1.5 equiv. of KBr give slightly improved yields. Terminal aliphatic olefins, e.g., 1-octene and butyl allyl ether need longer reaction times for complete conversion (Table 2, entries 13 and 14, 19 and 20). On the otherhand internal aliphatic olefins (5-decene, 2,3-dimethyl-2-butene) show a fast conversion of the olefin, but epoxide formation needs longer times compared to the aromatic olefins.
Conclusion
Table 1 - Epoxidation of alpha-methylstyrene using the NaOCl/KBr system
Table 2 - Epoxidation of various alkenes using NaOCl
{kind=link}
{kind=link}
In summary, we have shown that various non-activated olefins can be converted to epoxides by using simply sodium hypochlorite and bromide salt. It is surprising that this type of non-metal-catalyzed epoxidation has been previously largely overseen. Aromatic olefins furnish the corresponding epoxide with high selectivity at room temperature to 40°C in short time (<1-2h). Aliphatic olefins react somewhat more sluggishly. It is clear that the method described here is associated with the production of 1 equivalent of NaCl. Nevertheless, the procedure can be performed safely without any additional transition metals at ambient conditions. Further advantages of the procedure remain in the simplicity and the low-priced oxidant.
Experimental
General Information
All reactions were carried out without any special precautions under an atmosphere of air. Chemicals and solvents were purchased from Fluka and used as received. 1H and 13C NMR spectra were obtained on a Bruker ARX 400 spectrometer. Gas chromatographic analyses were run on a Hewlett-Packard GC6890 series, HP 5, 5% phenylmethyl siloxane, capillary (30m, 250µm, 0.25µm).
General Procedure
In a 100-mL Schlenk tube, KBr (357mg, 3.0 mmol or 47mg, 0.4 mmol, respectively), buffer (10mL, prepared by adjusting a 0.5 molar solution of KH2PO4 to a pH of 10.4 with a 2 molar NaOH solution), acetonitrile (10 mL), substrate (2.0 mmol) and diethylene glycol di-n-butyl ether (100µL, as internal standard for GC) were added. The reaction mixture was warmed to 40°C under 1000 rpm magnetic stirring using a thermostat. Aqueous NaOCl solution (Fluka commercial sodium hypochlorite, 1.1 mL of a 12.4% solution, d=1.2 g·mL-1, 1.1 equivalents) was added at once and stirring and temperature were maintained for 15 minutes to 24 hours depending on the substrate (see Tables above). Then, Na2SO3 (0.5 g) was added and the mixture was extracted with 20 mL of ethyl acetate. The combined organic layers were dried over MgSO4 and analyzed by GC.
For isolation of the product, the solvent was removed under vacuum and the crude epoxide was purified by column chromatography (hexane/ethyl acetate 10:1) or distillation.