Abstract
Isomerization of olefins, in particular the useful transformation of allyl to vinyl ethers is catalyzed by the hexaaquaruthenium(II) ion, producing the (E)-isomers under mild conditions.
Ring-opening metathesis polymerization of strained bicyclic olefins is catalyzed by [Ru(H2O)6]2+ under mild conditions1. Since solvents such as H2O or EtOH can be used2,3, this reaction is attractive with respect to ecological considerations. In analogy to Mo-, W-, or Ti-containing catalysts4,5, carbene intermediates have been postulated also for complexes of the iron triad. Experimental evidence for these species has recently been demonstrated for the case of ruthenium phosphines6. In the presence of [Ru(H2O)6]2+, acyclic olefins like allylic ethers and allylic alcohols do not undergo the expected metathesis reaction but show a migration of the C=C bond3. In this communication, we report on the range of olefin rearrangements catalyzed by [Ru(H2O)6]2+ and on the kinetics for specific examples.
Table.
Rearrangement Products
| Substratea | Productb |
| Hex-1-ene | (E)-Hex-2-ene (87) (E)-Hex-3-ene (13) |
| (Z)-Hex-2-ene | (Z)-Hex-2-ene (31) (E)-Hex-2-ene (31) (E)-Hex-3-ene (38) |
| (E)-Hex-2-ene | (E)-Hex-2-ene (59) (E)-Hex-3-ene (41) |
| (E)-Hex-3-ene | (E)-Hex-3-ene (90) (Z)-Hex-2-ene (10) |
| Hex-5-en-1-ol | Hex-5-ene-1-ol (15) Hex-4-ene-1-ol (85) |
| 4-Allyl-1,2-dimethoxybenzene | 3,4-Dimethoxy-1-propenylbenzene (>99) |
| 4-Allyl-2-methoxyphenol | 2-Methoxy-4-propenylphenol (>99) |
| Allyl phenyl ether | Phenyl propenyl ether (>99)d |
| Methylidenecyclopentanec | Methylidenecyclopentane (60) 1-Methylcyclopentene (40) |
Notes:
a) Acyclic olefins which showed no reaction, hex-5-en-2-on,
hept-6-enoic acid, 2-methylbut-1-ene, 2-methylbut-2-ene.
b) Percentage in brackets; determined by NMR.
c) Only successful
rearrangement with cyclic olefins. Nonreactive species: methylidene-
cyclohexane, methylidenecyclobutane, 4-vinylcyclohex-1-ene,
vinylcyclohexene.
d) Either (Z)- or (E)-isomer, 3Jolefin = 13 Hz.
Terminal olefins are converted in high yield into the thermodynamically more stable internal olefinsNote 1. The process is stereospecific producing the (E)-isomer (Table), except for (E)-hex-3-ene where a small amount of (Z)-hex-2-ene is also formedNote 2. We propose the C=C bond migration to proceed via formation of an olefin complex of Ru. Facile substitution of one or several H2O ligands by pi-acidic ligands is a well-documented reaction7,8. 4-Allyl-2-methoxyphenol is converted to 2-methoxy-4-propenylphenol by catalytic amounts of [Ru(H2O)6]2+. The concentration decrease of 4-allyl-2-methoxyphenol with time follows first-order behavior for as long as 50000 s at 31°CNote 3. This observation is a strong indication that the substitution of one H2O molecule of [Ru(H2O)6]2+ by 4-allyl-2-methoxyphenol is the rate-determining step. From the measurement of the temperature dependence of this reaction, the following kinetic parameters are derived: k298 (3.8 ± 0.1) 10-5 s-1, ΔH* = 44.7 ± 2 kJ·mol-1, and ΔS = -180.1 ± 6.5 J·K-1·mol-1. The quite large negative value of ΔS* indicates an associative pathway of the reaction. Further studies will deal with the reaction pathway for this isomerization process and its relation to olefin metathesis.
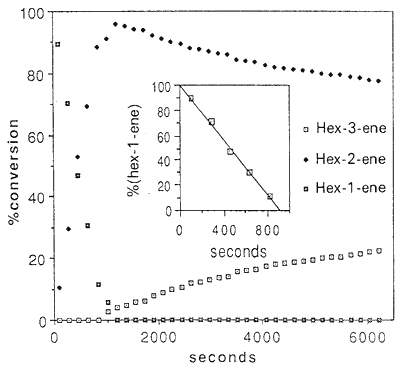
Figure. Time dependence of the concentration of hex-1-ene,
hex-2-ene and hex-3-ene in the isomerization with
[Ru(H2O)6(tos)2] as catalyst at 309 K in (D6)EtOH
The same reaction was studied for hex-1-ene. Its concentration decreases linearly with time up to 90% conversion. At this point, the subsequent formation of hex-3-ene begins (Fig.). Since hex-2-ene is the only detectable product at the beginning of the reaction. and since its concentration decreases upon formation of hex-3-ene. we conclude that the migration of the C=C bond occurs stepwise. i.e. hex-1-ene → hex-2-ene → hex-3-ene.
Rearrangement of allylic ethers and alcohols in aqueous solution3 with [Ru(H2O)6(tos)2] (tos = p-toluenesulfonate) also works in (D6)EtOH or in THF with [Ru(H2O)6(trif)2] (trif = trifluoromethanesulfonate)9 as catalysts: our experiments demonstrate that allyl phenyl ether is quantitatively converted to the phenyl vinyl ether at room temperature (12 h) with small amounts of catalyst. This opens applications in organic protecting-group chemistry. The base- and acid-stable allyl derivates of alcohols, acids, or carbonyl compounds can be converted to the corresponding acid-labile vinyl derivates on heating in presence of Pd/C10 or with tris(triphenylphosphane)rhodium(I) chloride at 60-80°C for several hours or leaving the mixture at room temperature for several days11. The exceedingly mild reaction conditions with [Ru(H2O)6(trif)2] as well as its solubility in a variety of organic solvents makes it an interesting catalyst for the cleavage of protecting groups via isomerization and acid-catalyzed hydrolysis.
Notes:
- For a typical experiment, 5 mg of [Ru(H2O)6]2+ was dissolved in 25 ml of D2O in a NMR tube and diluted with 400µl of (D6)EtOH. The olefin (100 µl) was injected, and the NMR tube was kept at 35°C for one day. Owing to the facile oxidation of [Ru(H2O)6]2+, all these operations were carried out under Ar.
- The configuration of the products was determined by observation of 3J olefinic coupling constants in 1H-NMR (3JZ, < 10, 3JE > 13 Hz) or by comparison with 13C-NMR spectra of the pure substances.
- A typical kinetic run consisted of the following, to 200 ul of a stock solution of 0.0453 M [Ru(H2O)6]2+ in a mixture of (D6)EtOH/D2O 15:1 was added 100 µl of olefin. The mixture was then diluted with (D6)EtOH to e total of 500 µl. and the NMR tube was placed into the thermostated compartment. All solvents were Ar-saturated, and the reactions took place in Ar-filled NMR tubes.