Abstract
The methylenation of a variety of catechols is described, employing cesium carbonate and bromochloromethane in dimethylformamide at 110°C or acetonitrile at reflux. The corresponding methylenedioxy derivatives are obtained in 86-97% yield. Utilization of this method provided β-hydrastine in 70% yield from its corresponding dihydroxy analog.
The methylenedioxy group is of special interest due to its occurrence in natural products1 and as a protecting group in organic synthesis2. However, its use has been limited due to the difficulty in formation of this ring system.
There have been several methods published in recent years describing improved methods for formation of methylenedioxy groups in the presences of a bronze3, nickel oxide4, or cupric oxide5 catalyst in dimethylsulfoxide (DMSO) or N,N-dimethylformamide (DMF). High yields of methylated catechols have also been obtained with excess potassium or cesium fluoride in DMF,6 with potassium carbonate in DMF,7 or with sodium hydride in hexamethylphosphoric triamide.8 Methylenedioxy groups have also been formed in the absence of a dipolar aprotic solvent by using a phase transfer catalyst at reflux.9 Although each of the methods cited has its merits, they afforded varying degrees of success when employed on the methylenation of 5,6-dihydroxy-1-tetralones 1 (R = H, F, or CH3). In fact, numerous attempts to methylenate 1 (R = F) under previous methodology3,5,6,8,9 provided the desired methylenedioxy derivative 2 (R = F) in low to moderate yield (10-50%).
Herein we wish to report a simple high yielding methylenation of catechols which requires neither strong base, special precautions, nor controlled addition of reagents. The reaction of catechol 1 (R = F) with bromochloromethane in DMF at 110°C in the presence of cesium carbonate provided the desired methylendioxy derivative 2 (R = F) in 86% yield.
Table I
Optimization of Methylenation of Catechol 1 (R = H)
Examplea |
X-CH2Cl [eq] |
M2CO3 [eq] |
Temp. | Time | Yield |
1 |
Br [1.5] | K [1.5] | 110°C |
2 h |
84% |
2 |
Br [1.5] | Cs [1.5] | 110°C |
2 h |
94% |
3 |
Br [1.5] | Cs [1.0] | 110°C |
2 h |
92% |
4 |
Br [1.5] | Cs [1.5] | 70°C |
5 h |
89% |
5 |
Cl [3.0] | Cs [1.5] | 110°C |
2.5 h |
82% |
6 |
Br [1.5] | Cs [1.5] | 82°C |
5 h |
95% |
a) Reactions were carried out on 0.5-1.0 g of 1 (R = H) at
a concentration of 0.4 M in anhyd. DMF, except Example 6
which was carried out in CH3CN.
Table I summarizes the optimization of this reaction on the readily available catechol 1 (R = H). Although potassium carbonate has been utilized as a base in this type of reaction,7 cesium carbonate provides higher and more consistent yields10 of methylenated product (Examples 1, 2). Also, less reactive catechols such as 1 (R = F) are methylenated in low to moderate yield when potassium carbonate is utilized as the base, as opposed to the high yield obtained with cesium carbonate.11 The reaction can also be conducted in acetonitrile in excellent yield, although the reaction time is increased due to the lower reaction temperature (Example 6). No intermolecular O-alkylation products were observed in either solvent. However, trace amounts of 1-hydroxy-2-methyl-5,6- methylenedioxy-naphthalene were occasionally observed, upon scale-up.
Table II
Methylenation of
Substituted Catechols
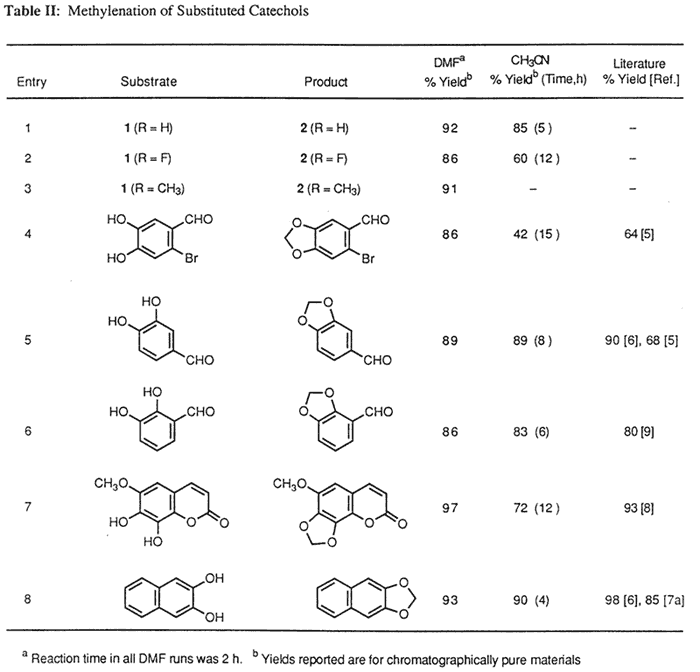
Table II contains a representative sampling of catechols that have been subjected to this method.12 The reaction can be carried out in either DMF or CH3CN, but higher yields are obtained in DMF when employing less reactive substrates. The increased effectiveness of DMF over CH3CN as solvent can be attributed in most cases to (a) the overall solubility of the starting material, (b) the solubility of the corresponding cesium phenoxides, and (c) the increased rate of reaction due to the elevated temperature obtainable in DMF. The reaction has also been extended to the preparation of (β-hydrastine 3 from the corresponding dihydroxy derivative 4.13 Previous attempts to methylenate this compound provided only decomposition products,6 whereas the present method provided (β-hydrastine in 70% after 4 hours at reflux in acetonitrile.
In summary, this method for the methylenation of catechols should find general utility, especially in cases where the base sensitivity of the catechol and/or product is a critical issue. Furthermore, the reaction works exceedingly well on catechols which are deactivated by neighboring electron withdrawing groups and/or ring deactivating groups.
Experimental
General Procedure for the Methylenation of Catechols in DMF
To a mechanically stirred degassed suspension of 5,6-dihydroxy-1-tetralone (9.6 g, 53.9 mmol) and Cs2CO3 (26.36 g, 80.9 mmol) in anhydrous DMF (130 mL) was added BrCH2Cl (5.26 mL, 80.9 mmol) and the resulting mixture was heated to 110°C. After 2 h, the reaction was cooled to room temperature and filtered through a pad of celite with EtOAc washing. The filtrate was concentrated almost to dryness and the residue diluted with water and extracted with EtOAc (3x). The extracts were combined washed with water, brine, dried over anhydrous MgSO4, filtered and concentrated to provide a tan solid. Chromatography (silica gel, elution with 10% EtOAc, hexanes) provided 9.4g (92%) of 5,6-methylenedioxy-1-tetralone as a white crystalline material, mp 134-136°C.
General Procedure for the Methylenation of Catechols in CH3CN
To a mechanically stirred degassed mixture of 5,6-dihydroxy-1-tetralone (3.9 g, 21.9 mmol) and Cs2CO3 (10.7 g, 32.9 mmol) in CH3CN (55 mL) was added BrCH2Cl (2.13 mL, 32.9 mmol) and the resulting suspension was heated to reflux. After 5 h, the reaction was cooled to RT and filtered through a pad of celite with EtOAc washing. The filtrate was concentrated and directly chromatographed on silica gel to provide 3.53 g (85%) of desired material.