Abstract
The allylation of pyrogallol 1-methyl ether (I) and the synthesis of myristicin from the pyrogallol 1-methyl 2-allyl ether (II; R = H) so formed, are described. The higher-boiling pyrogallol 1-methyl 3-allyl ether (III; R = H) also yields myristicin, apparently by migration of the allyl group to the meta-position.
Myristicin is the major constituent of the higher-boiling fractions of nutmeg and mace oils and it was shown by Thorns1 to be 1-methoxy-2,3-methylenedioxy-5-allylbenzene (V). This structure was confirmed by the synthesis of myristicinaldehyde2, and is now further confirmed by the synthesis of myristicin itself, a preliminary report of which was given in a letter to Nature3.
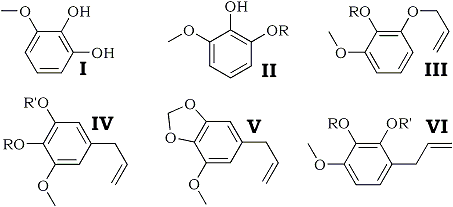
Allylation of pyrogallol 1-methyl ether (I)4 gave two monoallyl ethers differing in boiling point by 5°C. The lower-boiling ether was characterised by its 3,5-dinitrobenzoate and identified as pyrogallol 1-methyl 2-allyl ether (II; R = H) by methylation to the 1,3-dimethyl ether5,6 (II; R = Me) which was characterised by rearrangement to 2-hydroxy- 1,3-dimethoxy-5-allylbenzene (IV; R = H, R' = Me), identified as its 3,5-dinitrobenzoate.
Pyrolysis of (II; R = H) gave 2,3-dihydroxy-1-methoxy-5-allylbenzene (IV; R = R' = H), which formed a crystalline monoacetate and diacetate, and on treatment with methylene iodide and potassium carbonate, by the method of Perkin and Trikojus7, was converted into myristicin (V), identical with the natural product obtained from oil of nutmeg. This identity was established by the comparison of samples of tetrabromomyristicin, isomyristicin, and tetrabromoisomyristicin prepared from natural and synthetic myristicin.
The higher-boiling monoallyl ether was identified as pyrogallol 1-methyl 3-allyl ether (III; R = H) since on methylation and pyrolysis it yielded the same phenol (3,5-dinitrobenzoate as did pyrolysis of authentic 1,2-dimethoxy-3-allyloxybenzene (III; R = Me), prepared from pyrogallol 1,2-dimethyl ether4,8. On pyrolysis of (III ; R = H) and methylenation of the product, myristicin was again obtained, although in somewhat smaller yield than before. Methylation of the product of pyrolysis of (III; R = H) gave 3,4,5-trimethoxy-1-allylbenzene (IV; R = R' = Me), identified by conversion into 3,4,5-trimethoxybenzoic acid (O-trimethylgallic acid). Thus it appears that the allyl group has migrated to the meta-position in this pyrolysis. Pyrolysis of the methyl and ethyl ethers of (III; R = H), however, showed the normal migration of the allyl group to the ortho-position, yielding (VI ; R = Me, R' = H) and (VI; R = Et, R' = H) respectively.
Migration of the allyl group to the meta-position maybe largely influenced by the 2-hydroxyl group and may explain the formation of 1,2-dihydroxy-3,4-dimethoxy-5-allylbenzene exclusively in the pyrolysis of the crude monoallyl ether of 1,2-dihydroxy-3,4-dimethoxybenzene9.
Experimental
Allylation of Pyrogallol 1-Methyl Ether
Finely powdered, anhydrous potassium carbonate (18 g.) was suspended in acetone (20 ml.) in an atmosphere of hydrogen, and a solution of crystalline pyrogallol 1-methyl ether4,10 dissolved in acetone (40 ml.) slowly added. The potassium salt of the phenol crystallised and set to a solid mass but this gradually disintegrated on adding allyl bromide (16 g.) and boiling under reflux for 5 hours. After distillation of the acetone and dissolution of the solids in 2N-sulphuric acid (100 ml), the crude product was extracted twice with ether, and the phenols isolated by extraction of the ethereal solution with sodium hydroxide (8 g in all) in water (100 ml). The alkaline solution was immediately acidified with hydrochloric acid, and the liberated phenols removed by two extractions with chloroform. Unchanged pyrogallol 1-methyl ether was removed from the chloroform solution by shaking it with lead acetate solution (twice) until no more lead salt was precipitated. After removal of the lead salt by filtration, the chloroform solution was dried (Na2SO4), the chloroform evaporated, and some residual inorganic material removed by extraction with light petroleum (bp 40-60°C) and filtration of the solution. On removal of the petroleum the mixed monoallyl ethers (13.5 g) distilled at 0.30-0.35 mmHg/130°C (bath temp), as a colourless oil. Distillation through a 10 cm Widmer column gave two fractions: A, bp 98°C/0.45-0.42 mmHg, and B, bp 103-106°C/0.55-0.52 mmHg. These were refractionated with the same column, fraction B being added to the distillation flask after the first two fractions had been collected.
The fractions obtained were as follows:
- bp 92.5-94°C/0.25 mmHg (4 g) identified below as pyrogallol 1-methyl 2-allyl ether.
50 mg heated for 1 hour with 3,5-dinitrobenzoyl chloride (70 mg) and pyridine (0.5 ml) on the water-bath, kept at room temperature overnight, and then heated for a further 45 minutes, gave the 3,5-dinitrobenzoate as a pale yellow solid after being treated with dilute hydrochloric acid and washed with sodium carbonate solution and water. It formed colourless needles from 90% alcohol, mp 111-112°C. - bp 93-95°C/0.25 mmHg (1.6 g)
- bp 97.5-100°C/0.25 mmHg (2.1 g)
- bp 99-104°C/0.2 mmHg (4.4 g) identified below as pyrogallol 1-methyl 3-allyl ether.
Its 3,5-dinitrobenzoate, prepared as with the isomer above, crystallised as pale straw-coloured needles from alcohol, mp 133-134°C.
Added in Proof: Surrey's failure11 to separate or even to distil the monoallyl ethers of pyrogallol 1-methyl ether, without rearrangement, may be due to contamination of his material with pyrogallol 1-methyl ether or with oxidation products. When prepared and purified by the above method no difficulty was experienced in fractionating quantities up to 25g without rearrangement, although rearrangement was rapid at 170°C (bath temp).
Identification of Fraction 1
Fraction 1 (0.8 g) was methylated with methyl sulphate and 10% potassium hydroxide solution, the product extracted with ether, and the extract washed with dilute potassium hydroxide solution and with water and then dried and distilled in a vacuum, yielding a pale yellow oil (0.8 g), which was heated in an oil-bath till the temperature of the liquid rose above the temperature of the bath (at 225°C), heating being continued until the temperatures became equal again. On cooling, the product was dissolved in sodium hydroxide solution, and the solution washed with ether, acidified, and extracted with ether. After drying and removal of the ether, the residual yellow oil was converted into its 3,5-dinitrobenzoate, which formed canary-yellow aggregates of needles from alcohol, mp 129-130°C.
Authentic pyrogallol 1,3-dimethyl 2-allyl ether was prepared by the method of Mauthner5,6 and rearranged to 2-hydroxy-1,3-dimethoxy-5-allylbenzene, the 3,5-dinitrobenzoate of which formed identical canary-yellow aggregates from alcohol, mp 129-130°C, alone or mixed with the ester described above.
2,3-Dihydroxy-1-methoxy-5-allylbenzene (IV; R = R' = H).
Pyrogallol 1-methyl 2-allyl ether (Fraction 1; 2 g) was heated in an atmosphere of hydrogen to 170-175°C; the temperature of the liquid rose rapidly to 237°C, then fell, and was kept by external heating at 200°C for 1 minute. On distillation, 2,3-dihydroxy-1-methoxy-5-allylbenzene was obtained as a viscous oil, bp 112.5-115°C/0.22-0.30 mmHg (1.9 g), which on treatment with acetic anhydride and pyridine at room temperature for 24 hours, followed by washing with water and sodium hydrogen carbonate solution gave a monoacetate, colourless needles, mp 98-99°C, on crystallisation from dilute alcohol and then from ether–light petroleum. Acetylation of the 2,3-dihydroxy-1-methoxy-5-allylbenzene (0.3 g) with acetic anhydride (1.25 ml) and pyridine (1 ml) at 100°C for 1 hour, followed by standing overnight at room temperature, treatment with 2N-sulphuric acid and ether, and washing the ether solution with 2N-sulphuric acid and 2N-sodium carbonate, and drying, gave, on evaporation of the ether and treatment with light petroleum (bp 60-80°C; 3 ml), 2,3-diacetoxy-1-methoxy-5-allylbenzene, thick, colourless, pointed prisms from ether–light petroleum (bp 40-60°C), mp 75-76°C.
Myristicin (V)
2,2,3-Dihydroxy-1-methoxy-5-allylbenzene (2.9 g) was dissolved in acetone (15 ml), methylene iodide (4.3 g) and potassium carbonate (2.2 g) added, and the mixture refluxed on a boiling water-bath in an atmosphere of hydrogen. After 5 hours more potassium carbonate (2.2 g) and methylene iodide (1.1 g) were added, and refluxing continued for a further 2 hours. After removal of the acetone, ether and 2N sulphuric acid (25 ml) were added, and the ether solution was separated and washed with 2N-sodium hydroxide (20 ml. in all), dried, and distilled, yielding myristicin (1.03 g), bp 95–97°C/0.2 mmHg, nD20° 1.5426. This was characterised by treatment of 0.2g with bromine (0.3 ml), added dropwise below 0°C, the excess bromine being removed in a vacuum after a few minutes; the tetrabromo-derivative remained, and was washed with alcohol; it crystallised from alcohol as colourless needles (0.25 g), mp 128°C, identical with the tetrabromo-derivative obtained similarly from authentic myristicin, bp 148-151°C/16 mmHg, nD20° 1.5403, obtained by fractionation of oil of nutmeg.
Isomyristicin
Synthetic myristicin (0.2 g) was heated for 48 hours on the boiling water-bath with potassium hydroxide (powdered; 0.8 g) and alcohol (3 ml). On steam distillation the drops of oil obtained crystallised on cooling, and formed colourless needles from alcohol (50 mg), mp 43°C, alone or mixed with isomyristicin obtained by similar treatment of the natural product. Isomyristicin (natural or synthetic), on treatment with bromine as described for myristicin, gave the tetrabromo-derivative, mp 159°C, separately or mixed.
Identification of Fraction 4 (III; R = H)
(a) Methylation and pyrolysis
Fraction 4 (1.9g) was treated with methyl sulphate (3 lots of 2 ml. each) and shaken, with addition of 10% sodium hydroxide solution till alkaline, after each lot of methyl sulphate. The methyl ether was then isolated by extraction with ether, the extract being washed with 10% sodium hydroxide and water, dried, and the ether evaporated. The residual oil was then rearranged by being heated to 235°C for 15 minutes, during which time the temperature in the liquid rose to 3°C below that of the bath temperature, then fell and remained steady 5°C below the bath temperature. The product was distilled, bp 95-100°C/0.12mmHg (1.0g) dissolved in ether, and extracted with 10% sodium hydroxide, the extract acidified, and the phenolic product again extracted with ether and again distilled. It was characterised by preparation of the 3,5-dinitrobenzoate by heating the oil (0.2 g.), 3,5-dinitrobenzoyl chloride (0.3 g), and pyridine (1 ml) for 1 hour at 100°C. On cooling, adding 2N sulphuric acid and ether, washing the ether solution with acid, water, 2N sodium hydroxide and water again, drying, and evaporating the ether, the crude ester crystallised (0.14 g.), mp 115-116°C, and formed pale yellow needles from alcohol, mp 118-119.5°C.
(b) Pyrolysis of authentic pyrogallol 1,2-dimethyl 3-allyl ether
The required ether (III; R = Me) was prepared by allylation of pyrogallol 1,2-dimethyl ether4,8 (4.1 g.) dissolved in acetone (25 ml.) with allyl bromide (3.5 ml.) and potassium carbonate (dry, sieved; 4.1 g.) by refluxing for 6 hours. The mixture was then diluted with water, the acetone distilled, and the product isolated by extraction with ether, the extract being washed with 10% sodium hydroxide and water, dried, the ether removed, and the residual oil distilled, bp 98-103°C/0.2mmHg (3.8 g).
Pyrolysis of pyrogallol 1,2-dimethyl 3-allyl ether (2.1 g) followed the same course as described above for the methyl ether of Fraction 4, except that more heat was evolved and the temperature of the liquid rose to 5° above the bath temperature. The pyrolysis product (oriented by the methods described below as 3-hydroxy-1,2-dimethoxy-4-allylbenzene) was distilled, bp 103-105°C/0.25 mmHg, and 0.2 g. converted into the 3,5-dinitrobenzoate as described above. The crude ester (0.46g), mp 116-117°C, formed pale yellow needles from alcohol, mp 118-119.5°C, alone or mixed with the material derived from Fraction 4.
(c) Orientation of the 3-hydroxy-1,2-dimethoxy-(x)-allylbenzene
(i) The 3-hydroxy-1,2-dimethoxy-(x)-allylbenzene obtained above (5 g.) was methylated with methyl sulphate (2 lots of 5 ml.) and 10% sodium hydroxide, the mixture being finally heated to 100° for 10 minutes and the methyl ether isolated by extraction with ether. The allyl group was then isomerised by treatment of the methyl ether (4.2 g) with potassium hydroxide (15.6 g) in alcohol (60 ml) for 48 hours on the steam-bath, the product being isolated by steam distillation and extraction of the distillate with ether (3.5 g), and then oxidised by adding potassium permanganate (powdered; 7 g) gradually with shaking to the solution in acetone (140 ml). Heat was evolved, and the reaction was completed by warming the mixture for a few minutes on the steam-bath. Excess of permanganate and manganese dioxide were then reduced by passing sulphur dioxide into the solution. The oxidation product separated and formed colourless needles from water (0.5 g), mp 99-102°C, undepressed by admixture with 2,3,4-trimethoxybenzoic acid of similar melting point.
(ii) The above 3-hydroxy-1,2-dimethoxy-(x)-allylbenzene (7.5 g.) was dissolved in 10% sodium hydroxide (300 ml) and heated to the boiling point during the gradual addition of ethyl sulphate (32 ml; 30 minutes). After 2 hours' refluxing the ethyl ether was isolated by extraction with ether, and the recovered phenol again treated, using the same procedure and combining the products. On distillation, the product was obtained as a pale yellow oil, bp 150-152°C/22 mmHg (7.5 g), identified below as chiefly 1,2-dimethoxy-3-ethoxy-4-allylbenzene
The allyl group was isomerised by treatment of 6 g. with potassium hydroxide (26 g) in alcohol (100 ml) for 48 hours on the steam-bath, the propenyl compound being isolated by steam distillation and extraction of the distillate with ether. 1,2-Dimethoxy-3-ethoxy-4-propenylbenzene was obtained as a colourless oil, bp 163-167°C/24 mmHg (4.9 g).
The constitution of this product (4.0g) was shown by oxidation with potassium permanganate (8 g) in acetone (160 ml) as described above for the trimethoxy-compound. The acid oxidation product in this case separated as an oil which crystallised on cooling (1.5g) and was purified by crystallisation first from dilute alcohol (charcoal) and then thrice from dilute methyl alcohol; it formed colourless needles, mp 79-81°C, identical with authentic 3,4-dimethoxy-2-ethoxybenzoic acid (Manske, Ledingham, and Holmes, loc. cit.)
Pyrolysis of Pyrogallol 1-Methyl 3-Allyl Ether (III; R = H)
Fraction 4 (4 g) was heated in an atmosphere of hydrogen; rearrangement took place at ca. 190°C, the temperature of the liquid rising above the bath temperature. After 15 minutes' heating the product was distilled as a colourless, viscous oil, bp 114-116°C/0.2 mmHg (3.5 g).
Myristicin from the pyrolysis product
The above pyrolysis product (3.3 g.) was methylenated by the method described in the previously mentioned preparation of myristicin, and the neutral product distilled, bp 86-87°C/0.20-0.18 mmHg (0.6 g). On treatment of 0.18 g with bromine (3 ml) at 0°C, it formed tetrabromomyristicin (0.25 g), colourless needles from alcohol, mp 128°C, undepressed by admixture with an authentic specimen.
The identity of the methylenated material with myristicin was further confirmed by rearrangement to isomyristicin, and the formation of its tetrabromo-derivative, mp 157°C, raised to 158°C on admixture with authentic material, mp 159°C.
Identification of the pyrolysis product
The above pyrolysis product (0.8 g) was treated with methyl sulphate (3 lots of 1 ml) in an atmosphere of hydrogen, with addition of 10% sodium hydroxide and shaking till alkaline after each lot of methyl sulphate. The methyl ether (0.7 g) was isolated by extraction with ether and rearranged by being heated for 48 hours on the steam-bath with potassium hydroxide (2.6g) and alcohol (10 ml). Steam distillation and ether extraction of the distillate yielded an oil (0.5g) which was dissolved in acetone (20 ml.) and oxidised by the gradual addition of potassium permanganate (powdered; 1g) with shaking. Heat was evolved, and the reaction was completed by warming on the steam-bath for a few minutes. Any unchanged oil was removed by steam distillation and sodium hydrogen sulphite and dilute sulphuric acid were added to the residual solution. The product and which crystallised (0.15 g.) had mp 105-145°C and formed colourless needles after one crystallisation from water (charcoal); mp 169-170°C, undepressed by admixture with 3,4,5-trimethoxybenzoic acid (O-trimethylgallic acid).