Myristicin (I) has been converted into homomyristicinic acid (VII) by oxidation with potassium permanganate and by a multistep procedure2, based on the ozonolysis of the double bond of II, followed by oxime formation and subsequent dehydration to VI. However, the reported melting points of the final products are different, 127°C1 and 108°C2 respectively.
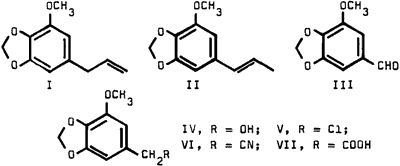
We wish to report a modified synthesis of homomyristicinic acid (VII) as shown in the above sequence of reactions. This procedure, which afforded the low melting point product, circumvents the disadvantage of the erratic yields in the ozonolysis step2 and renders large scale synthesis feasible.
The starting material (I), obtained by distillation of nutmeg oil, could be readily converted to the stable isomyristicin (II) by a facile tautomeric rearrangement. Double bond fission of II was accomplished in 48% yield, via hydroxylation followed by vanadium pentoxide oxidation.3 The reduction of aldehyde III to alcohol IV with sodium borohydride proceeded smoothly in nearly quantitative yield. Treatment of IV with thionyl chloride afforded the chloro derivative V in 88% yield.
The attempted conversion of V to the cyanide VI by conventional methods4 failed. However, treatment with sodium cyanide in dimethylsulfoxide readily afforded the desired cyanide in excellent yield; it had the spectral characteristics expected for VI. Basic hydrolysis of VI gave homomyristicinic acid (VII) with the same melting point as that reported by Hahn and Schales.2
Experimental
5-Allyl-1-methoxy-2,3-methylenedioxybenzene (Myristicin)(I)
Myristicin (I) was obtained by distillation of commercial nutmeg oil (Naarden) in 5% yield, bp 148-150°C/15 mm, lit.5,6 bp 149.5°C/15 mm. Bromination of I with bromine in ethanol gave 2,6-dibromo-5-methoxy-3,4-methylenedioxy-1-(2',3'-dibromo)propylbenzene, mp 127-129°C (ethanol), lit.5 mp 130°C.
1-Methoxy-2,3-methylenedioxy-5-propenylbenzene (Isomyristicin) (II)
A mixture of I (50 g), sodium hydroxide (27.5 g) and ethanol (160 ml) was boiled at gentle reflux (24 hrs). After evaporation of the reaction mixture in vacuo, the solid residue was dissolved in water (400 ml) and extracted with ether (3x100 ml). The ethereal extract was dried and evaporated to dryness. Distillation of the residue at reduced pressure afforded 41 g (83%) of II, bp. 166°C/18 mm. Recrystallized from methanol gave white needles, mp 42-44°C, lit.5 mp 44-45°C.
Bromination of II with bromine in ethanol gave 2,6-dibromo-5-methoxy-3,4-methylenedioxy- 1-(1',2'-dibromopropyl)benzene, mp 156-157°C (ethanol), lit.5 mp 156°C.
3-Methoxy-4,5-methylenedioxybenzaldehyde (III)
To a mixture of II (40 g) and 6% hydrogen peroxide in tert-butyl alcohol (360 ml), vanadium pentoxide (0.7 g) was added. The reaction mixture was set aside at room temperature for 2 hrs. Initial evolution of gas (foaming) was controlled by external cooling. On standing, the cooled solution deposited yellow crystals, which were collected. The crude product was dissolved in 10% aqueous sodium acid sulphite (900 ml), filtered and the filtrate made alkaline with 10% sodium hydroxide. The precipitate was collected, washed with water and crystallized from ethanol to give 18 g (48%) of pale yellow needles of III, mp. 130-131°C, lit.8 mp 130-131°C.
3-Methoxy-4,5-methylenedioxybenzylalcohol (IV)
Sodium borohydride was added portionwise to a solution of III (11.4 g) in methanol (160 ml). The mixture was boiled to reflux (1 hr). The solvent was removed and the residue was taken up in a mixture of ether:water (1:1) (200 ml). The aqueous layer was separated and extracted with ether. The combined ethereal extracts were washed and dried. The solvent was removed at reduced pressure to afford the crude alcohol. Recrystallization from toluene gave 10.8 g (95%) of IV, mp. 65-66°C.
3-Methoxy-4,5-methylenedioxybenzyl Chloride (V)
A solution of thionyl chloride (7 ml) in dry benzene (17 ml) was added dropwise and with stirring to a solution of IV (11 g) in dry benzene (110 ml). Stirring was continued at room temperature for 1.5 hr. and then boiled to reflux for 10 min. The solvent was evaporated to dryness and the crystalline residue suspended in water (100 ml) and extracted with ether. The ethereal extracts were washed with 5% aqueous potassium carbonate, dried and evaporated. The crude product was recrystallized from acetone to give 10 g (83%) of white needles of V, mp 82-83°C.
(3-Methoxy-4,5-methylenedioxy)phenylacetonitrile (VI)
A solution of V (11 g) in DMSO (50 ml) was added with stirring to a solution of sodium cyanide (4.9 g) in DMSO (200 ml); stirring was continued for 6-hrs. The mixture was poured into water (600 ml) and the precipitate collected. The crude product, crystallized from methanol, gave 9.3 g (97%) of VI, mp 89-90°C, lit.2 mp 90°C.
(5-Methoxy-4,5-methylenedioxy)phenylacetic acid (Homomyristicinic acid) (VII)
A mixture of VI (9 g), sodium hydroxide (4 g), water (11 ml) and ethanol (11 ml) was boiled for 7 hrs. The resulting solution was diluted with water (20 ml) and acidified with 6N hydrochloric acid (30 ml). The precipitate was collected, washed with water and recrystallized from water to give 8 g (81%) of white needles of VII, mp 107-108°C, lit. 108°C2 and 127°C1.