Abstract:
The nitro group in aliphatic and aromatic nitro compounds also containing reducible substituents such as ethene, nitrile, acid, phenol, halogen, ester, etc., are selectively and rapidly reduced at room temperature to corresponding amines in good yields by employing hydrazinium monoformate, in the presence of commercial zinc dust. It was observed that, hydrazinium monoformate is more effective than hydrazine or formic acid and reduction of nitro group occurs without hydrogenolysis in the low cost zinc dust compared to expensive metals like palladium.
Rapid and selective reduction of nitro compounds is of importance for the preparation of amino derivatives in the organic synthesis both practically and industrially, particularly when a molecule has other reducible moieties.1–5 The synthesis and biological evaluation of aromatic amines are also active and most important areas of research and their chemistry by derivative formation is widely studied. (6,7) Numerous new reagents have been developed for the reduction of aromatic nitrocompounds. (8) Though some of these are widely used, still they have limitations based on safety and handling considerations. For example, catalytic transfer hydrogenation9–11 of nitro or azido compounds in the presence of metals such as palladium on carbon or platinum on carbon require stringent precautions, because of their flammable nature in the presence of air. In addition, some methods require compressed hydrogen gas, which is highly diffusible and flammable, and vacuum pump to create high pressure within reaction flask. To overcome these difficulties, several new methodologies have also been developed.12,13 However, little attention has been given to the reduction of aliphatic nitrocompounds, 14–16 which are traditionally reduced by high-pressure catalytic hydrogenation.17–19 In addition to above mentioned limitations, most of these methods are unfortunately subject to substantial limitations as concerns the reducible functionalities and lack therefore desired generality for the true synthetic utility. Moreover, poor selectivity was reported in the reduction of aromatic nitro compounds, which have halogen, nitrile, carboxyl, hydroxyl etc., as substituents. Reduction at reflux temperature for hours together can cause rearrangements and cyclization in poly functional nitrocompounds. Therefore, we examined several methods to improve reduction process, and especially to obtain selectivity over reducible or other labile substituents. In this context, use of 5% platinum on carbon was found to be efficient but not cost effective.20
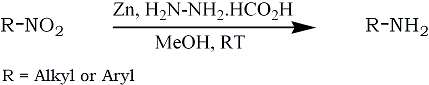
Recently, metal mediated reactions have been found to have wide scope in organic synthesis, because of their simple work-up and selectivity. Several methods have been developed based on the use of a variety of metals such as magnesium,21 indium,8,22,23 tin,24 zinc.25 The utility of zinc for the synthesis of β-α-unsaturated ketones by a reaction of an acid chloride with allyl bromide26 and homoallylic alcohols27 has been demonstrated. Further, the zinc mediated preparation of triphenyl phosphonium ylides,28 Fridel–Crafts acylation20 and carbamates formation30 has been demonstrated.
In this communication, we wish to report, a rapid and simple reduction of aliphatic and aromatic nitrocompounds to the corresponding amino derivatives by using commercial zinc dust and hydrazinium monoformate, at room temperature (Scheme 1). This new system reduced with ease a wide variety of nitro compounds directly to the corresponding amines and many functional groups are tolerated. This system is not helpful to obtain directly an amino carbonyl compound, due to the formation of hydrazone derivative with donor. However, the nitro hydrazones are reduced to corresponding amino hydrazones by this system. But, the thing is, in order to get amino carbonyl derivative, hydrazone should be subjected to hydrolysis. In both nitro aldehydes as well as nitro ketones, the products were obtained in almost pure and comparable yields.
The reduction of nitrocompound in the presence of zinc dust and hydrazinium monoformate was complete within one to ten min. The course of reaction was monitored by thin layer chromatography and IR spectra. The work-up and isolation of the products were easy. Thus, all the compounds reduced (Table 1) by this system were obtained in good yields (90–95%). All the products were characterized by comparison of their TLC, IR spectra, and melting points with authentic samples. A control experiment was carried out using nitro compounds with hydrazinium monoformate but without zinc dust, does not yield the desired product. No other intermediates, such as nitroso or hydroxylamine could be detected in the reaction mixture. In order to test the selectivity, reduction was attempted with p-dichlorobenzene, p-chloro-m-cresol, β-naphthol, cinnamic acid, acetanilide, benzoic acid, anisole, benzonitrile, phenyl acetate, etc., at laboratory temperature. However, the reaction failed to give any reduced product.
| Nitro compound | Reaction time (min) |
Product | Yielda | mp (Found) | mp (Lit) |
| o-Nitrophenol | 2 | o-Aminophenol | 93% | 173–175°C | 174°C |
| m-Nitrophenol | 2 | m-Aminophenol | 94% | 121–123°C | 123°C |
| 2,4-Dinitrophenol | 2 | 2,4-Diaminophenol | 95% | 79–80°C | 79°C |
| o-Nitrotoluene | 2.5 | o-Toluidineb | 93% | 142–144°C | 144°C |
| m-Nitrotoluene | 2.5 | m-Toluidineb | 92% | 124–126°C | 125°C |
| p-Nitrotoluene | 2 | p-Toluidine | 94% | 44–45°C | 45°C |
| 2,4-Dinitrotoluene | 2.5 | 2,4-Diaminotoluene | 91% | 98–99°C | 99°C |
| o-Dinitrobenzene | 3 | o-Phenylenediamine | 93% | 101–104°C | 102°C |
| m-Dinitrobenzene | 2.5 | m-Phenylenediamine | 93% | 63–65°C | 64°C |
| a-Nitronaphthalene | 2 | a-Naphthylamine | 92% | 50–51°C | 50°C |
| β-Nitronaphthalene | 2 | β-Naphthylamine | 94% | 111–113°C | 113°C |
| o-Nitroanisole | 2.5 | o-Anisidineb | 94% | 58–60°C | 60°C |
| m-Nitroanisole | 2 | m-Anisidinec | 95% | 81–82°C | 80°C |
| p-Nitroanisole | 2 | p-Anisidine | 95% | 56–57°C | 57°C |
| o-Nitroaniline | 2.5 | o-Phenylenediamine | 93% | 100–103°C | 102°C |
| m-Nitroaniline | 2.5 | m-Phenylenediamine | 94% | 64–65°C | 64°C |
| p-Nitroaniline | 2 | p-Phenylenediamine | 94% | 140–143°C | 141°C |
| m-Nitrobenzyl alcohol | 3 | m-Aminobenzyl alcohol | 91% | 96–98°C | 97°C |
| p-Nitrobenzamide | 2.5 | p-Aminobenzamide | 92% | 115–116°C | 114°C |
| p-Nitrophenylacetate | 2.5 | p-Aminophenylacetatec | 93% | 148–151°C | 150°C |
| 2,2'-Dinitrodibenzyl | 5 | 2,2'-Diaminodibenzyl | 90% | 222–226°C | 223°C |
| o-Nitrobenzoic acid | 2 | o-Aminobenzoic acid | 92% | 144–147°C | 145°C |
| m-Nitrobenzoic acid | 2.5 | m-Aminobenzoic acid | 94% | 174–176°C | 174°C |
| p-Nitrobenzoic acid | 2.5 | p-Aminobenzoic acid | 85% | 184–187°C | 186°C |
| o-Nitrochloro benzene | 2 | o-Chloroanilineb | 92% | 99–100°C | 99°C |
| m-Nitrochloro benzene | 2.5 | m-Chloroanilineb | 92% | 120–123°C | 122°C |
| p-Nitrochloro benzene | 2 | p-Chloroanilne | 93% | 70–71°C | 71°C |
| o-Nitrobromo benzene | 2.5 | o-Bromoanilineb | 94% | 115–117°C | 116°C |
| m-Nitrobromo benzene | 3 | m-Bromoanilineb | 94% | 118–121°C | 120°C |
| p-Nitrobromo benzene | 2.5 | p-Bromoaniline | 95% | 65–66°C | 66°C |
| m-Nitroiodo benzene | 2.5 | m-Iodoaniline | 91% | 119–121°C | 119°C |
| p-Nitrocinnamic acid | 3 | p-Aminocinnamic acidd | 90% | 265–268°C | 265–270°C |
| p-Nitrobenzonitrile | 2.5 | p-Aminobenzonitrile | 91% | 84–85°C | 83–85°C |
| p-Nitrophenylacetonitrile | 3 | p-Aminophenylacetonitrile | 91% | 45–48°C | 45–48°C |
| p-Nitrophenethyl alcohol | 3 | p-Aminophenethyl alcohol | 92% | 108–111°C | 108–110°C |
| p-Nitroacetanilide | 3 | p-Aminoacetanilide | 93% | 163–165°C | 163°C |
| 3,5-Dinitrobenzoic acid | 4 | 3,5-Diaminobenzoic acid | 91% | 234–238°C | 235–238°C |
| Methyl-p-nitrocinnamate | 3.5 | Methyl-p-aminocinnamate | 91% | 128–130°C | 129°C |
| Nitromethane | 2 | Methylamined | 80% | 230–233°C | 232–234°C |
| Nitroethane | 2 | Ethylamined | 81% | 106–108°C | 107–108°C |
| 1-Nitropropane | 2 | 1-Aminopropaned | 84% | 158–160°C | 160–162°C |
| 1-Nitrobutane | 2.5 | 1-Aminobutanee | 75% | 78–80°C | 78°C |
- a) Isolated yields are based on single experiment and the yields were not optimized.
- b) Isolated as benzoyl derivative.
- c) Isolated as acetyl derivative.
- d) Isolated as hydrochloride salt.
- e) Boiling point.
Hydrazinium diformate, a white crystalline compound obtained by the neutralization of one mole of hydrazine hydrate with two mole of 85% formic acid was found to be inactive to this system. Further, hydrazinium monoformate is more effective than either hydrazine or formic acid with zinc dust. Most of the reactions are complete in less than one minute as monitored by the disappearance of the starting materials and concomitant formation of the product via TLC methods.
Thus the reduction of nitrocompounds can be accomplished with commercial zinc dust instead of expensive platinum or palladium etc., with out effecting the reduction of any reducible or hydrogenolysable substituents. The yields are virtually quantitative and analytically pure. The obvious advantages of proposed method over previous methods are: (i) selective reduction of nitro compounds, in the presence of other reducible or hydrogenolysable groups, (ii) easy to operate, (iii) rapid reduction, (iv) high yields of substituted amines, (v) avoidance of strong acid media, (vi) no requirement of pressure apparatus, and (vii) less expensive. This procedure will therefore be of general use, especially in the cases where rapid, mild and selective reduction is required. Further investigations of other useful applications related to deblocking of protecting groups in peptide synthesis are in progress.
Typical Procedure
The hydrazinium monoformate was prepared by neutralizing slowly, equal moles of hydrazine hydrate and 85% formic acid in an ice water bath, with constant stirring. Thus obtained hydrazinium monoformate solution is used as such for reduction. A suspension of an appropriate nitrocompound (5 mmol) and zinc dust (10 mmol) in methanol or in any suitable solvent (3 mL) was stirred under nitrogen atmosphere with hydrazinium monoformate (2 mL), at room temperature. The reaction was exothermic and effervescent. After the completion of reaction (monitored by TLC), the reaction mixture was filtered through celite. The organic layer is evaporated and the residue was dissolved in chloroform or dichloromethane or ether was washed with saturated sodium chloride solution to remove excess of hydrazinium monoformate. The organic layer after drying and evaporation gave the desired amino derivative.
In order to get good yield of volatile aliphatic amine, the reaction was carried out by controlled addition of hydrazinium monoformate, through the top of ice water circulated condenser and by immersing the reaction flask in a cold-water bath. After filtration, whole reaction mixture was neutralized with HCl. The solvent was evaporated under reduced pressure. The residue was lyophilized or subjected to column chromatography. Aliphatic amines are obtained as their hydrochloride salts up to 80% yield.