Abstract
Various aldoximes and ketoximes were selectively reduced to the corresponding amines by catalytic transfer hydrogenation employing low cost magnesium powder and ammonium formate at room temperature. Many other functionalities such as halogens, -OH, -OCH3, -COOH and -CH3 remained unaffected. The hydrogenation is fast, mild, clean, cost effective and high yielding.
Introduction
The conversion of carbonyl derivatives to amines via oximes is a useful transformation in the synthesis of numerous organic compounds and key intermediates in the biosynthesis of many pharmacological important compounds.1,2 In comparison, the corresponding reactions of C=N bonds have been studied much less. Although the addition to the C=N bond of imines, hydrazones and oximes are well known,3 the development of such reaction is often limited by the poor electrophilicity of the C=N carbon atom. Numerous new reagents have been developed for the reduction of oximes to amines including NaBH4,3,4 LiAlH45,6 and indium/acetic acid.7 Though some of these are widely used, still they have limitations based on chemoselectivity and economic considerations. Catalytic hydrogenation is also commonly used,8,9 although the success of reaction is sensitive towards catalyst, solvent and substrate. Further, catalytic hydrogenation employs highly diffusible, low molcular weight, flammable hydrogen gas and requires pressure equipment. Electrolytic reduction of oximes to amines, in acid solution is also reported,10 but this system offers very low yield.
There has been growing interest in the use of metals in synthetic chemistry. Zinc,11 indium,12 tin,13 magnesium14-18 have been used in the synthesis of many organic compounds. The utility of magnesium for the synthesis of pinacols,14 silaspiro compounds15 has been demonstrated. Further, the magnesium-mediated reduction of aromatic nitro compounds,16 Barbier reaction17 and in situ Grignard reactions18 has also been demonstrated. The application of ammonium formate19,20 in the field of catalytic transfer hydrogenation for the reduction of variety of organic compounds and in the synthesis of peptides is peer reviewed.
Nowadays, the heterogeneous catalytic transfer hydrogenation method has proved to be a potent choice for reduction of organic compounds over traditional hydrogenation or other methods of reduction as it involves mild reaction condition, easy work-up and high degree of selectivity.19-24 Earlier reports reveal that catalytic transfer hydrogenation of oximes to amines had been achieved with systems like ammonium formate/10% Pd/C,23 cyclohexene/10% Pd/C.24 But these systems require long reaction times as long as 5–10 hours at reflux temperature, expensive catalyst and also offer very low yield. Moreover, stringent precautions must be taken while employing palladium on carbon because of its flammable nature in presence of air.
Scheme 1.
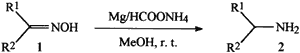
R1, R2 = H, alkyl, aryl
Recently, we have explored the utility of magnesium powder and hydrazinium monoformate; a new hydrogen source for deblocking some commonly used protecting groups in peptide synthesis.25 Herein we report a rapid, selective and simple reduction of oximes to the corresponding primary amines by using low cost magnesium powder and ammonium formate at room temperature as depicted in the Scheme 1. Various other functionalities like halogens, -OH, -OCH3, -COOH and –CH3 are tolerated.
Table 1.
Mg-catalyzed reduction of oximes to amines using HCOONH4
| 1/2 | R1 |
R2 |
Time |
Yielda |
B.P |
B.P. (Ref) |
a | Ph | H | 38 |
91% |
182-184°C |
185°C26 |
b | Me | H | 40 |
66%b |
166°Cc |
|
c | Me | Me | 42 |
64%b |
152°Cc |
|
d | Ph | Me | 40 |
89% |
184°C |
184-6°C27 |
e | Ph | Ph | 45 |
93% |
295-296°C |
295°C26 |
f |
p-OH-Ph |
Me | 52 |
92% |
123°Cc |
|
g |
p-MeO-Ph |
H | 55 |
85% |
236°C |
236-7°C26 |
h |
3,4,5-MeO-Ph |
H | 52 |
84% |
122°C |
121°C26 |
i |
p-Cl-Ph |
H | 32 |
94% |
212-214°C |
215°C26 |
j | 50 |
81% |
157-159°C |
158-60°C26 |
||
k | 55 |
79% |
135°C |
134°C26 |
||
l | 52 |
88% | 100°C |
101°C26 |
Notes:
- Isolated yields. Unoptimized, based on a single experiment.
- Low yield due to low bp of the product, isolated as HCl salt.
- Melting point.
- 1j: Succinaldehyde dioxime, 2j: 1,4-Diaminobutane
- 1k: Cyclohexanone Oxime, 2k: Cyclohexylamine
- 1l: p-Nitro-Benzaldoxime, 2l: p-Amino-Benzylamine
The results given in the Table 1 reveal the viability of using Mg/HCOONH4 system for the reduction of oximes. The course of reaction was monitored by TLC and IR spectra. The work-up and isolation of the products were easy. Thus, the oximes reduced (few examples are listed in the Table 1) by this system were obtained in good yield. The products were characterized by comparison of their boiling points or melting points, TLC and IR spectra with authentic samples. The disappearance of strong absorption bands between 1690–1640 cm-1 due to C=N stretching and between 3650–3500 cm-1 due to O-H stretching and appearance of two strong absorption bands between 3500–3300 cm-1 of -NH2 group clearly show that the oximes were reduced to corresponding primary amines.
A control experiment was carried out using oximes with ammonium formate, but without magnesium powder does not yield any reduced product and the starting material is recovered in 100%. This confirms the role of magnesium as catalyst. The reaction was also carried out for several hours with ammonium chloride instead of ammonium formate expecting an efficient reduction, but the yield is very low. Further, we observed that in the case of nitro oximes, the nitro group at aryl residue and also oxime group underwent reduction to yield respective diamine product at room temperature.
In summary, the reduction of oximes can be accomplished in a short time with magnesium powder instead of expensive catalyst like palladium23,24 at room temperature and many other functionalities are tolerated. The yields are almost quantitative and the compounds analytically pure. This magnesium-catalyzed procedure provides a very efficient, inexpensive, mild, and general methodology for reduction of oximes to amines.
Experimental
Materials
The oximes were either commercially available or prepared from the corresponding carbonyl compound by standard methods. In cases where the oxime was obtained as an E/Z-mixture, no attempts were made to separate such mixtures and they are used as such for the reduction. Magnesium powder was purchased from SISCO Research Laboratories Pvt. Ltd., Bombay (India) and was treated with 0.01 N hydrochloric acid for about 2 min. It was filtered through a sintered glass funnel and washed with water, dry methanol and dry ether. Thus obtained magnesium was vacuum dried and stored. Ammonium formate and 60–120-mesh silica gel (for column chromatography) were purchased from E. Merck (India) Ltd. All of the solvents used were analytical grade or were purified according to standard procedures. Thin layer chromatography was carried out on silica gel plates obtained from Whatman Inc. The melting points were determined by a Thomas–Hoover melting point apparatus and are uncorrected. IR spectra were recorded on a Shimadzu FTIR-8300 spectrometer.
Typical Procedure
To a solution of the substrate (10 mmol) in methanol or in any other suitable solvent (20 mmol) was added ammonium formate (30 mmol) and magnesium powder (1g, 0.041 mol). The mixture was stirred under nitrogen atmosphere at room temperature. The reaction was exothermic and effervescent. After the completion of reaction (monitored by TLC), the reaction mixture was filtered through celite. The organic layer is evaporated and the residue was dissolved in chloroform or dichloromethane or ether and washed with saturated sodium chloride solution to remove excess ammonium formate. The organic layer was dried over anhydrous sodium sulphate and evaporation of the organic layer followed by purification either by preparative TLC or by column chromatography to yield the desired product.
In order to obtain the volatile aliphatic amines in good yield, the reaction was carried out using a condenser cooled with ice water and the reaction flask immersed in a cold-water bath. After filtration, the reaction mixture was neutralized with HCl. The solvent was evaporated under reduced pressure. The residue was lyophilized or subjected to column chromatography by using 60–120 mesh silica gel and a suitable eluting system <50:50 chloroform:benzene (for entry 2e), 60:40 chloroform:benzene (for entries 2a & 2d), 80:20 chloroform:benzene (for entry 2j), 90:10 chloroform:benzene (for entry 2k), 80:20 chloroform:methanol (for entries 2b & 2c), 85:15 chloroform:methanol (for entries 2f & 2g), 90:10 chloroform:methanol (for entry 2i), 95:5 chloroform:methanol (for entries 2h & 2l)>. Aliphatic amines were obtained as their hydrochloride salts in up to 65% yield.