Methaqualone (Quaalude) Synthesisby RhodiumCheapskate's excellent step-by-step quaalude synthesis guide MaDMAx' scaleup of Cheapskate's above procedure A Survey of Reported Syntheses of Methaqualone (Quaalude) and its Isomers (PDF) IntroductionMethaqualone, also known as 3,4-dihydro-2-methyl-4-oxo-3-o-tolylquinazoline, 2-methyl-3-(2-methylphenyl)-4- (3H)-quinazolinone, Metholquizolone, QZ-2, 2-Methyl-3-o-tolyl-4(3H)-quinazolinone, RIC-272, TR-495 and Quaalude. Methaqualone was introduced as an anxiolytic (Quaalude, Sopor) in 1965 as safe barbiturate substitutes. Experience showed, however, that their addiction liability and the severity of withdrawal symptoms were similar to those of barbiturates. By 1972, "luding out," taking methaqualone with wine, was a popular college pastime. Excessive use leads to tolerance, dependence and withdrawal symptoms similar to those of barbiturates. Overdose by glutethimide and methaqualone is more difficult to treat than barbiturate overdose, and deaths have frequently occurred. In the United States, the marketing of methaqualone pharmaceutical products stopped in 1984 and methaqualone was transferred to Schedule I of the CSA. The LD50 is for mice 1250 mg/kg and for rats 326 mg/kg (Strasenburg Labs), also given as 255 mg/kg for rats (Goldenthal). The human dose (as sedative) is set to 150 mg (A.S. Paterson). Methaqualone freebase, mw 250.30, mp 113-115°C. Soluble in ethanol, ether and CHCl3. Insol. H2O. Methaqualone HCl, mw 285.80, mp 235-237°C. Soluble in ethanol and ether. Insol. H2O. 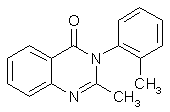
MethaqualoneFrom Anthranilic Acid (Klosa)[2]This is an improved version of Klosas original synthesis of methaqualone. The method first devised by him used phosphorous pentachloride instead of polyphosphoric acid as condensing agent, which during the reaction produced clouds of corrosive HCl gas [1]. 7g Anthranilic Acid, 5ml o-Toluidine and 4 ml glacial acetic acid was mixed in a round-bottomed flask. To this mixture was slowly added 40-50 grams of polyphosphoric acid, and the mixture was heated during 20-30 minutes to 140-160°C. After this, the mixture was heated to 180°C for 10 minutes, then cooled and poured in 150-200ml water, and neutralized with 20% Na2CO3 solution. Methanol was added until a lasting turbidity became present in the solution, and after one hour, the free base Methaqualone precipitated, mp 111-113°C, and was once more recrystallized in this fashion, mp 113-115°C. Yield 55%. From Anthranilic Acid [5,6]This method is similar to the one used by Klosa, but this one drives out the water byproduct from the reaction mixture with heat, instead of letting polyphosphoric acid bind it. This method also manages to push the yield a little further. The product formed in the first step is N-acetylanthranilic acid, which can be made as described in the precursors section below. If so, begin with the addition of o-Toluidine to the already made N-acetylanthranilic acid. Anthranilic acid (10 grams) is dissolved in acetic anhydride (20 ml) and the temperature raised progressively to 190-200°C at which temperature distillation takes place. The last traces of acetic acid are removed under vacuum and after cooling to about 50-60°C, o-toluidine (10 grams) is added in portions. The temperature is then raised to 170-200°C when the excess water and o-toluidine is gradually distilled off, finally maintaining the temperature at 180-200°C for 2 hours. After cooling to about 100°C, 30ml dilute HCl is added and the mixture boiled and stirred. The solution is neutralized with NaOH with stirring, and the crude product which separates is recrystallized twice from alcohol. The yield is 70% of theory. From N-acetylanthranilic Acid [5]This method is similar to Klosa's first synthesis of methaqualone, the difference being that this one uses POCl3 instead of PCl5, and uses ready made N-acetylanthranilic acid. By performing the reaction in toluene, the yield is very good. In spite of this, the method is best left alone, as the POCl3 will release lots of HCl gas during decomposition. o-Toluidine (10 grams) is mixed with a solution of N-acetylanthranilic acid (20 grams) in toluene (30 grams) in a vessel equipped with a stirrer and means of cooling. A solution of phosphorus oxychloride (10 grams) in toluene (30 grams) is added dropwise with stirring and then the temperature is raised to the boiling point for two hours with further stirring. After cooling, the precipitate so formed is filtered off, dried and dissolved in boiling dilute HCl. On cooling and making alkaline with NaOH, a viscous oil separates which crystallises after a few hours. The crystals are collected the next day and purified by recrystallisation from alcohol to yield about 80% of the theoretical quantity of methaqualone freebase (m.p. 114-115°C). The salts my be obtained by the addition of the appropriate acids to the base formed above. From Isatoic Anhydride (Manhas)[3]Method A A mixture of isatoic anhydride (1.6 g, 0.01 mole) and o-toluidine (1,1 g, 0,01 mol) is heated to 120° for 2 hours. The reaction mixture after cooling is triturated with ether (or dissolve the brown mixture in warm acetone and add water to crash out the crystals). The resulting solid is collected by suction and recrystallized from a 50:50 mixture of dichloromethane and petroleum ether to give the intermediate aminoamide (2,2'-dimethylbenzanilide): yield: 1,7 g (75%): m.p. 110°. 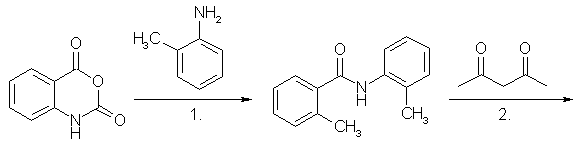
1. Isatoic anhydride condenses with o-Toluidine to give 2,2'-dimethylbenzanilide 2. 2,2'-Dimethylbenzanilide condenses with acetylacetone to form Methaqualone (see structure above) A mixture of 2,2'-dimethylbenzanilide (0.5 g, 0.0025 mole), acetylacetone (0.39g, 0.0025 mol) in ethanol (30 ml) containing a few drops of concentrated hydrochloric acid is refluxed for 1 h. On cooling the title compound separated as the hydrochloride salt: yield: 0,59 g, (85%), m.p. 235-237°. Method B A mixture of 8g isatoic anhydride and 5.5g o-toluidine in toluene (500-750 ml) is refluxed for two hours. Acetylacetone (2.5g) containing a few ml of concentrated hydrochloric acid (to form the hydrochloride) is then added, and refluxing is continued for another hour. Evaporation of the solvent gives methaqualone hydrochloride, which is purified by recrystallization from hot methanol. The yield is 80%. PrecursorsAnthranilic Acid (2-aminobenzoic acid)Method 1: Anthranilic Acid from o-ToluidineN-acetyl-o-Toluidine[4] 50ml o-Toluidine is placed in a 250ml round-bottomed flask containing a few boiling stones, and a reflux condenser is attached. Through an addition funnel placed in a side-arm of the flask, or possibly through the condenser, 50ml of acetic anhydride is added pretty rapidly, and the solution will begin to boil. Maintain the boiling for five minutes with external heating, and quickly pour the reaction contents in a beaker containing 1000ml of cold water. A precipitate of pinkish needles will form, which are filtered off with suction and is purified by dissolving in 25ml of boiling methanol, cooling the solution, and filtering off the beautiful glistening white needles which precipitates. The crystals are air dried, and weighs approximately 50 grams. Do not leave the crystals too long in the air, and keep them in an air-tight container, because they sublime readily at room temperature. N-Acetyl-Anthranilic Acid [7] Dissolve 20 grams of N-acetyl-o-Toluidine and 50 grams of magnesium sulfate in 2500ml water with stirring and heat to 80°C. Add 60 grams of finely powdered potassium permanganate crystals and increase the temperature to 85°C, and continue to stir for 2 hours. Add 50 ml of alcohol to decompose excess permanganate, and filter the solution HOT with suction. The N-Acetyl-Anthranilic Acid is precipitated from the solution with the addition of dilute sulfuric acid. It is purified by dissolving in dilute NaOH solution, and reprecipitating with dilute H2SO4. The yield after air drying is approximately 18 grams. This N-Acetyl-Anthranilic Acid can be molarly substituted for the Anthranilic Acid in the Klosa synthesis above (No 3), with omitting of the acetic acid. Anthranilic Acid [7] 10 grams of N-Acetyl-Anthranilic Acid is added to 100ml concentrated HCl, and boiled under reflux for 1-2 hours. The solution is cooled, and the crystals of anthranilic acid is filtered off and recrystallized from hot water. The approximate yield after air drying is XX g. Method 2: Anthranilic Acid from phtalic anhydride Phtalimide (Method 1) [4] Place 100 g. of phthalic anhydride and 106 ml. of concentrated ammonia in a 1-litre round-bottomed flask fitted with a wide air condenser or a liebig condenser (without cooling water) with an inner diameter of more than 10 mm. Heat first over a wire gauze and then over a free flame until the mixture is in a state of quiet fusion and forms a homogeneous melt (the temperature reaches 300° in about 1.5-2 hours; all the water is evaporated during the first hour). Shake the flask occasionally during the heating and push down any material which sublimes into the condenser with a glass rod. Pour the contents of the flask whilst still hot into a porcelain basin or casserole, allow to cool, and grind to a fine powder in a mortar. The phthalimide (95 g.)is practically pure and melts at 233-234°C. It may be recrystallised from ethanol or methanol (see method B). Phtalimide (Method 2) [4] Intimately mix 99g of pure phthalic anhydride and 20g of urea, and place the mixture in a 1 litre long-necked, round-bottomed flask. Heat the flask in an oil bath at 130-136°C. When the contents have melted, effervescence commences and gradually increases in vigour: after 10-20 minutes, the mixture suddenly froths up to about three times the original volume (this is accompanied by a rise in temperature to 160-160°) and becomes almost solid. Remove the flame from beneath the bath and allow to cool. Add about 80 ml. of water to disintegrate the solid in the flask, filter at the pump, wash with a little water, and then dry at 100°. The yield of phthalimide, m.p. 233° (it is practically pure) is 86 g. If desired, the phthalimide may be recrystallized from 1200 ml. of methylated spirit; the first crop consists of 34 g. of m.p. 234°, but further quantities may be recovered from the mother liquor. Anthranilic Acid (Method 2) [8] Anthranilic Acid from phtalimide, by Methaco(s)mic Anthranilic Acid (Method 2) [4] Prepare a solution of 30 g. of sodium hydroxide in 120 ml. of water in a 360 ml. conical flask and cool to 0° or below in a bath of ice and salt. Add 26.2g (8.4 ml.) of bromine in one portion and shake (or stir) until all the bromine has reacted. The temperature will rise somewhat; cool again to 0° or below. Meanwhile, prepare a solution of 22g. of sodium hydroxide in 80 ml. of water. Add 24 g. of finely- powdered phthalimide in one portion to the cold sodium hypobromite solution; stir vigorously while swirling the contents of the flask and add the prepared sodium hydroxide solution rapidly. The solid will dissolve and the temperature will rise to about 70°. Warm the mixture to 80° for about 2 minutes. Filter, if necessary. Cool in ice and add concentrated hydrochloric acid slowly and with stirring until the solution is just neutral (about 60 ml. are required). [It is recommended that a little of the alkaline solution be set aside in case too much acid is added.] Precipitate the anthranilic acid completely by the gradual addition of glacial acetic acid (20-25 ml) are required): it is advisable to transfer the mixture to a 1 litre beaker as some foaming occurs. Filter off the acid at the pump and wash with a little cold water. Recrystallise from hot water with the addition of a little decolourising carbon; collect the acid on a Buchner funnel and dry at 100°. The yield of pure anthranilic acid, m.p. 145°, is 14 g. Acetylacetone (2,5-pentanedione)Acetylacetone from Acetone and Ethylacetate Claisen Condensation of acetone with ethyl acetate in presence of NaOCH3 yields Acetylacetone. o-Toluidine (2-aminotoluene)o-Toluidine from Toluene Nitrotoluene Toluene is nitrated with a HNO3/H2SO4 mixture to yield o- and p-nitrotoluene. Toluidine [7] This should be performed in a strong, easily cleaned vessel, equipped with a powerful stirring device and a reflux condenser. This reaction can be modified to use tin instead of iron. 60 cc of water and 120 g of fine, cleaned, iron powder are placed in the reaction vessel and vigorous stirring is begun. The vessel contents are heated to 90-95°C and 10 cc of hydrochloric acid is poured in. 100 g of nitrotoluene (mono-nitrotoluene) is now added, at such a rate as to hold the temp at exactly 100°C (a few ml at a time). After the addition is complete, the temp will have to be maintained at 100°C with the heating device until the smell of nitrotoluene is gone. Vigorous stirring must be used through the entire operation. Set the reflux condenser to distill and lead steam directly into the vessel to steam distill the toluidine out of the reaction contents. If available, use only o-nitrotoluene as substrate in this step, to save chemicals and the separation step can be omitted. o-Toluidine [7] The crude oily toluidine mixture from the above steam distillation is separated from the water, ice and salt are added to the oil, and the mixture is stirred. A whitish-yellow crystalline compound appears, which is the hydrate of p-toluidine. This is filtered off through an ice cooled filter funnel, and then is well pressed to remove any oily o-toluidine. The o-toluidine is separated from the filtrate with a separation funnel. This crystallization should preferably be repeated to ensure that all p-toluidine is removed. References[1] J. Klosa, Synthese von Chinazolon-4-derivaten J. Prakt. Chem. 14, 84-98 (1961)[2] J. Klosa, Notiz ueber Synthese von Chinazolon-4-derivaten, J. Prakt. Chem. 20, 283-4 (1963) [3] Manhas, A Facile Synthesis of Methaqualone and Analogs, Synthesis 5, 309-10 (1977) [4] A. I. Vogel, Textbook of Practical Organic Chemistry, 3rd Ed. [5] Sittig & Marshal, Pharmaceutical Manufacturing Encyclopedia, Vol 2, p 969, 2nd Ed. [6] Brit. Pat. 843,073 [7] P. Buzz, Recreational Drugs, Loompanics (1989) [8] C. Graebe, Ueber die Hofmann'sche Reaction, Ber. 35, 2747-2752 (1902) |