

#137 MP
METAPROSCALINE; 3,4-DIMETHOXY-5-(n)-PROPOXYPHENETHYLAMINE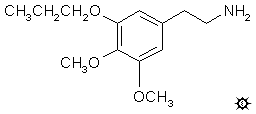
|
| [3D .mol structure] |
A mixture of 38.7 g 3-bromo-4,5-dimethoxybenzaldehyde and 17.2 g cyclohexylamine was heated with an open flame at about 120 °C until it appeared to be free of H2O. The residue was put under a vacuum (0.2 mm/Hg) and distilled at 146-160 °C yielding 44.6 g 3-bromo-N-cyclohexyl-4,5-dimethoxybenzylidenimine as a clear oil which did not crystallize. The imine stretch in the infra-red was at 1640 cm-1. Anal. (C15H20BrNO2) C,H.
A solution of 31.6 g 3-bromo-N-cyclohexyl-4,5-dimethoxybenzylidenimine in 300 mL anhydrous Et2O was placed in an atmosphere of He, stirred magnetically, and cooled with an dry ice/acetone bath. Then 71 mL of a 1.55 M solution of butyllithium in hexane was added over a 2 min period. The reaction mixture turned cloudy and a light precipitate formed which seemed heaviest at the half-way point. Stirring remained easy and was continued for 10 min. There was then added 35 mL of butyl borate at one time. The precipitate dissolved, and the stirred solution allowed to return to room temperature. There was then added 200 mL of an aqueous solution containing 20 g ammonium sulfate. The Et2O layer was separated, washed with saturated ammonium sulfate solution, and the organic solvents removed under vacuum. The residue was dissolved in 250 mL of 70% MeOH and 14 mL of 30% hydrogen peroxide added in small portions. This reaction was very exothermic, and stirring was continued for 1 h. The reaction mixture was then added to 500 mL H2O, which knocked out white solids. A small sample of this intermediate, N-cyclohexyl-3,4-dimethoxy-5-hydroxybenzylidineimine was recrystallized from MeOH to a white crystal with a mp of 148-149 °C and which showed the C=N bond as a doublet at 1635 and 1645 cm-1 in the infra-red. These wet solids were suspended in 200 mL 5% HCl and heated on the steam bath for 1 h. Stirring was continued until the reaction was again at room temperature and then it was extracted with 2x100 mL CH2Cl2. These extracts were pooled and in turn extracted with 2x75 mL dilute NaOH. The aqueous extracts were reacidified with HCl, and reextracted with 2x100 mL CH2Cl2. These extracts were pooled, and the solvent removed under vacuum to yield a brown viscous oil as a residue. This was distilled at 105-120 °C at 0.2 mm/Hg to yield 8.8 g of 3,4-dimethoxy-5-hydroxybenzaldehyde as a distillate that set to white crystals. Recrystallization from toluene/hexane gave a sample with the mp 64-65 °C. The literature mps are several, ranging from at about 60 °C to about 70 °C.
A solution of 4.7 g of 3,4-dimethoxy-5-hydroxybenzaldehyde in 75 mL acetone was treated with 6.0 g powdered KI, 16 mL (21 g) propyl bromide, and 7.0 g finely powdered anhydrous K2CO3, and this mixture was held at reflux on a steam bath for 15 h. The reaction mixture was added to 1 L H2O, made strongly basic, and extracted with 3x100 mL CH2Cl2. The extracts were pooled, washed with 5% NaOH, and the solvent removed under vacuum yielding 8.8 g of a yellow oil, undoubtedly containing propyl iodide. This residue was distilled at 133-145 °C at 0.15 mm/Hg to yield 4.5 g of 3,4-dimethoxy-5-(n)-propoxybenzaldehyde as a white oil which did not crystallize. There was an appreciable pot residue. This product was clearly impure, having a minor, slower moving component not the starting phenol, as seen by TLC (on silica gel, with CH2Cl2 as a developing solvent). Fusion of a small amount of impure aldehyde with p-anisidine produced a crystalline anil which, on hydrolysis with dilute acid, produced an aldehyde sample free of this impurity. But as this sample also remained as an oil, the above crude product was used in the following preparation.
To a solution of 3.8 g 3,4-dimethoxy-5-(n)-propoxybenzaldehyde in 50 mL nitromethane, there was added 0.5 g anhydrous ammonium acetate. This was held at reflux for 50 min. The excess nitromethane was removed under vacuum and 2 volumes of boiling MeOH were added to the residue. The hot solution was decanted from some residual insolubles, and on cooling spontaneously crystallized. These solids were removed by filtration, washed sparingly with MeOH and air dried yielding 3.3 g yellow crystals of 3,4-dimethoxy-beta-nitro-5-(n)-propoxynitrostyrene as yellow crystals melting at 79-81 °C. Recrystallization from MeOH or cyclohexane neither improved the mp nor freed the product from a residual opalescenceseen in the melt. Anal. (C13H17NO5) C,H.
A solution of 1.5 g LAH in 30 mL anhydrous THF under He was cooled to 0 °C and vigorously stirred. There was added, dropwise, 1.0 mL of 100% H2SO4, followed by the dropwise addition of a solution of 2.3 g 3,4-dimethoxy-beta-nitro-5-(n)-propoxynitrostyrene in 10 mL anhydrous THF, over the course of 5 min. The mixture was stirred at 0 °C for a while, and then brought to a reflux on the steam bath. After cooling again, the excess hydride was destroyed with IPA added dropwise, followed by the addition of about 10 mL of 10% NaOH which was sufficient to covert the solids to a white, granular form. These were removed by filtration, the filter cake washed with IPA, the mother liquor and filtrates were combined, and the solvents were removed under vacuum to yield an amber oil. This residue was added to 75 mL dilute H2SO4 which produced a gummy insoluble phase which was physically removed with a spatula. The aqueous phase was washed with 3x50 mL CH2Cl2. It was then made basic with 25% NaOH, and extracted with 2x75 mL CH2Cl2. The solvent was removed from these pooled extracts and the residue distilled at 106-116 °C at 0.2 mm/Hg to provide 1.3 g of the product as a colorless liquid. This was dissolved in 4 mL IPA, neutralized with about 20 drops of concentrated HCl, and diluted with 4 volumes of anhydrous Et2O added slowly with continuous stirring. A white crystalline salt crystallized out spontaneously and was isolated by filtration, washed first with IPA, then with Et2O, and air dried giving 1.3 g 3,4-dimethoxy-5-(n)-propoxyphenethylamine hydrochloride (MP) with a mp of 170-171 °C. Anal. (C13H22ClNO3) C,H.
DOSAGE: greater than 240 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 160 mg) There might have been some disturbance at the three to four hour point, but it was extremely light if at all.
(with 240 mg) No effects whatsoever.
EXTENSIONS AND EXTRAPOLATIONS: The loss of activity on lengthening the carbon chain on the meta-oxygen from two to three (from metaescaline to metaproscaline) discouraged any further exploration at this specific point of the molecule. The isopropyl analog (3,4-dimethoxy-5-(i)-propoxyphenethylamine, metaisoproscaline, MIP) was started and carried along as far as the aldehyde, and abandoned with the discovery that metaproscaline was without activity. There were other fish to fry.
| [ |
[Main Index] | [Forward |